- Article
- Source: Campus Sanofi
- 23 Oct 2025
An introduction to Gaucher Disease
.jpg)
What is gaucher?
Gaucher disease is one of the most common lysosomal storage disorders, affecting an estimated 1 in 40,000 to 1 in 100,000 people around the world.1 It can be diagnosed at any age from infancy to late adulthood. It is an inherited deficiency of the lysosomal enzyme acid-β-glucosidase (glucocerebrosidase, GBA1), which results in the accumulation of glucocerebroside within lysosomes of macrophages.1
Gaucher disease can be classified into three types, which make a phenotypic continuum ranging from mild to severe nervous symptoms.2–4 The classic categories of types 1, 2, and 3 have blurred edges along the continuum of the disease.

Adapted from Sidransky E, 2004. Mol Genet Metab. 83(1–2):6–15.3
The table summarises aspects of Gaucher disease according to the three different types.1,5
| Type 1 Non neuronopathic(≥1/100 to <1/10) | Type 2 Acute neuronopathic (≥1/100 to <1/10) | Type 3 Chronic neuronopathic (≥1/100 to <1/10) | |
| Prevalence | 1:50,000 – 1:100,000 (pan-ethnic) 1:850 (Ashkenazi Jews) | < 1:150,000 (pan-ethnic) | < 1:150,000 (pan-ethnic) |
| Age at presentation | Any | Infancy | Childhood |
| Lifespan | Variable | < 2 y | < 40 y |
| Primary CNS disease | None | Severe | Mild to severe |
| Hepatosplenomegaly | Mild to severe | Severe | Mild to severe |
| Haematologic abnormalities | Mild to severe | Severe | Mild to severe |
| Osseous symptoms | Mild to severe | None | Mild to moderate |
References
- Mistry PK, et al. Am J Hematol 2011. 86(1):110–115.
- Charrow J, et al. Clin Genet 2007. 71(3):211–215.
- Sidransky E, et al. Mol Genet Metab. 2004. 83(1–2):6–15.
- Sidransky E, et al. Gaucher disease clinical presentation. Updated November
- Niederau C. Gaucher Disease 3rd edition. Bremen: Uni-Med, 2017.
Epidemiology of Gaucher disease
Although considered a rare disease in the general population, the prevalence of Gaucher disease is significantly higher in specific patient groups.1
Think Gaucher disease when considering the following:
-
Patients presenting with splenomegaly and/or thrombocytopenia
-
Patients with family members affected by Gaucher disease
-
Patients of Ashkenazi Jewish descent
Although the disease can affect anyone of any ethnicity, there is a high prevalence of Gaucher disease in certain ethnic backgrounds. Notably those of Ashkenazi Jewish descent have a disease prevalence of approximately 1 in 800.2
Approximately 1 in every 15 people of Ashkenazi descent is a carrier of Type 1 Gaucher disease.1
Therefore it is important to test close family of patients from this background.

As an autosomal recessive disorder, a carrier of Gaucher disease has a 50% chance of having a child which carries the Gaucher gene. If both parents are carriers, there is a 25% chance that the child will be affected with Gaucher disease. If your patient has an immediate relative who has been diagnosed with Gaucher disease, it is important to order a blood test straight away.


References
- Motta I, et al. Eur J of Haematol 2015. 96:352–359.
- Mistry PK, et al. Am J Hematol 2011. 86(1):110–115.
Signs and Symptoms
The signs of Gaucher disease may look similar to other haematological malignancies. Rule out Gaucher disease in patients who present with these symptoms.
Four key symptoms to look out for are:1
- Splenomegaly
- Thrombocytopenia
- Hepatomegaly
- Bone pain
What to look out for
The journey to diagnosis can be a challenge. However, there are some key symptoms and signs to look out for:2,3



.2025-07-31-15-39-59.png)

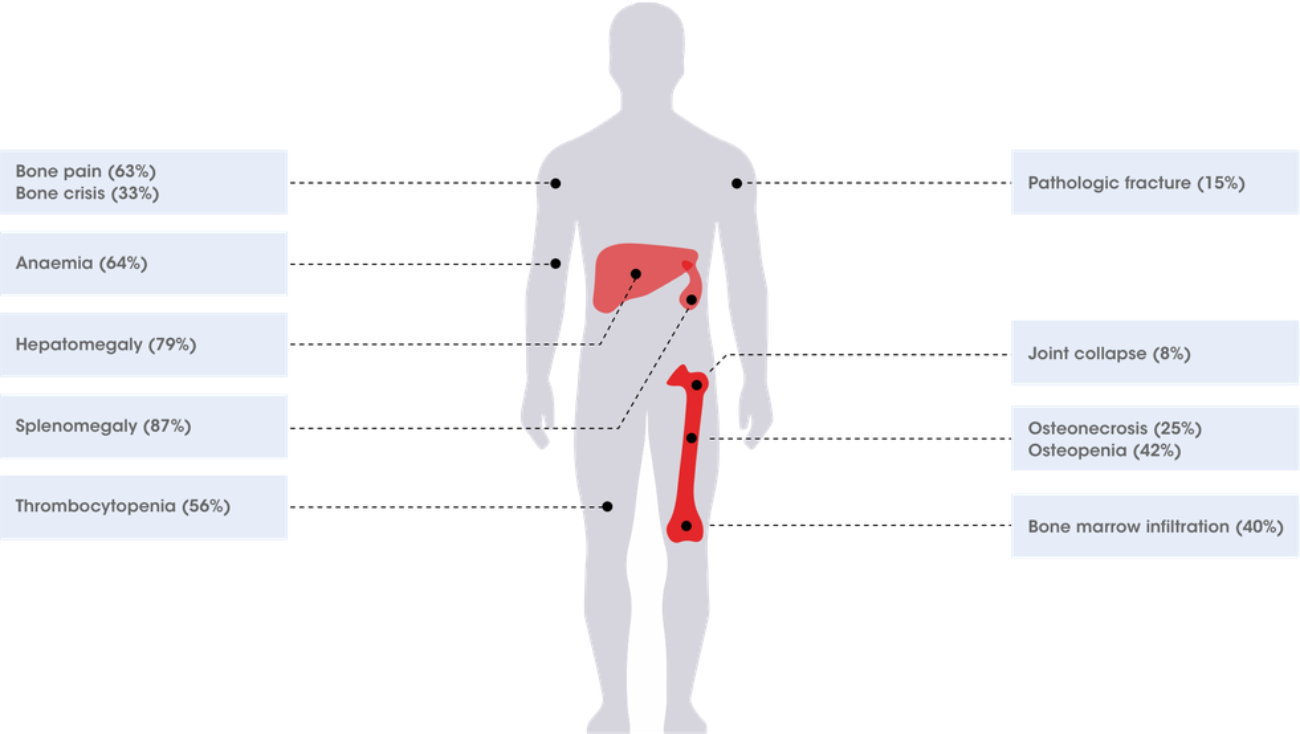
Gaucher symptoms prevalence data from Charrow J et al. Arch Intern Med. 2000. 160:2835–2843.3
When considering patients with these symptoms, where malignancy has been ruled out, think Gaucher disease.3–20
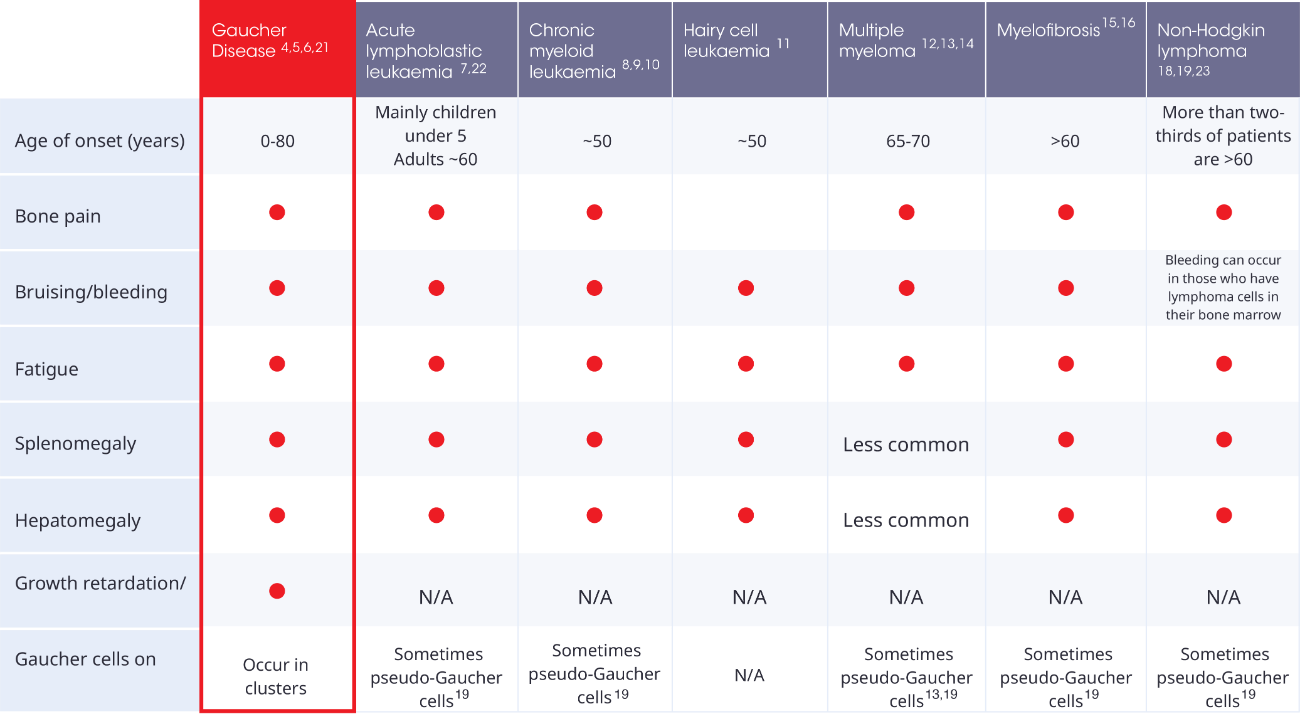
References
- Motta I, et al. Eur J of Haematol 2015. 96:352–359.
- Nagral A. J Clin Exp Hep 2014. 4(1):37–50
- Charrow J et al. Arch Intern Med. 2000. 160:2835-2843
- Mistry PK, et al. Am J Hematol. 2011. 86(1):110–115.
- Gaucher registry annual report. Genzyme Corp. 2006.
- Grabowski GA, et al. (2013). 'Gaucher Disease' in Valle DL, et al., OMMBID. McGraw-Hill Medical. DOI: 10.1036/ommbid.419
- Carrington PA, et al. J Clin Pathol. 1992. 45(4):360.
- Sawyers CL. N Engl J Med. 1999. 340(17):1330–1340.
- Savage DG, et al. Br J Hematol. 1997. 96(1):111–116.
- Hoffman R, et al. Hematology: basic principles and practice, 6th edition. Philadelphia, PA: Saunders, an imprint of Elsevier inc. 2013.
- Hairy cell leukaemia facts (FS16) Leukemia and Lymphoma Society. White Plains. NY. October 2013
- Al Farsi K. Oman Med J. 2013. 28(1):3–11.
- Scullin DC Jr, et al. Am J Med. 1979. 67(2):347–352.
- Multiple Myeloma. NHS. https://www.nhs.uk/conditions/multiple-myeloma/symptoms/. Accessed November 2025.
- Myelofibrosis facts (FS14). White Plains, NY. October 2013
- Tefferi A. Am J Hematol. 2013. 88(2):141–150.
- National Cancer Institute website. Adult Non-Hodgkin Lymphoma Treatment. https://www.cancer.gov/types/lymphoma/patient/adult-nhl-treatment-pdq. Accessed November 2025.
- Shankland KR, et al. Lancet. 2012. 380(9844):848–857.
- Lang E, et al. Diagn Cytopathol. 1999. 20:379–381
- Alterini R, et al. Haematologica. 1996. 81:282–283.
- Symptoms and signs of Gaucher disease. National Gaucher Foundation. Available at: https://www.gaucherdisease.org/about-gaucher-disease/symptoms/. Accessed November 2025.
- O'Donnell M. http://cancernetwork.com/cancer-management/acute-leukemias. Accessed November 2025.
- Non-Hodgkin Lymphoma - Symptoms. Cancer Research UK. https://www.cancerresearchuk.org/about-cancer/non-hodgkin-lymphoma/symptoms. Accessed November 2025.
MAT-XU-2504633 (v1.0) Date of preparation: November 2025