- Article
- Source: Campus Sanofi
- 16 Jul 2024
What is Pompe disease?
About Pompe disease
Pompe disease is a rare, progressive, and potentially fatal neuromuscular disease that affects multiple organ systems in the body.3
Also known as acid alpha-glucosidase deficiency, acid maltase deficiency (AMD), and glycogen storage disease type II (GSD-II), Pompe disease is a type of lysosomal storage disease and is characterised by the progressive accumulation of glycogen in muscle cells.2,3
An estimated 1 in 40,000 people worldwide are living with Pompe disease 4–6
What causes Pompe disease?
Pompe disease is a genetic disease, inherited in an autosomal recessive manner, and
is caused by dysfunctional or deficient acid alpha-glucosidase (GAA).2,3 This is the
enzyme responsible for breaking down glycogen in the lysosomes of cells.2
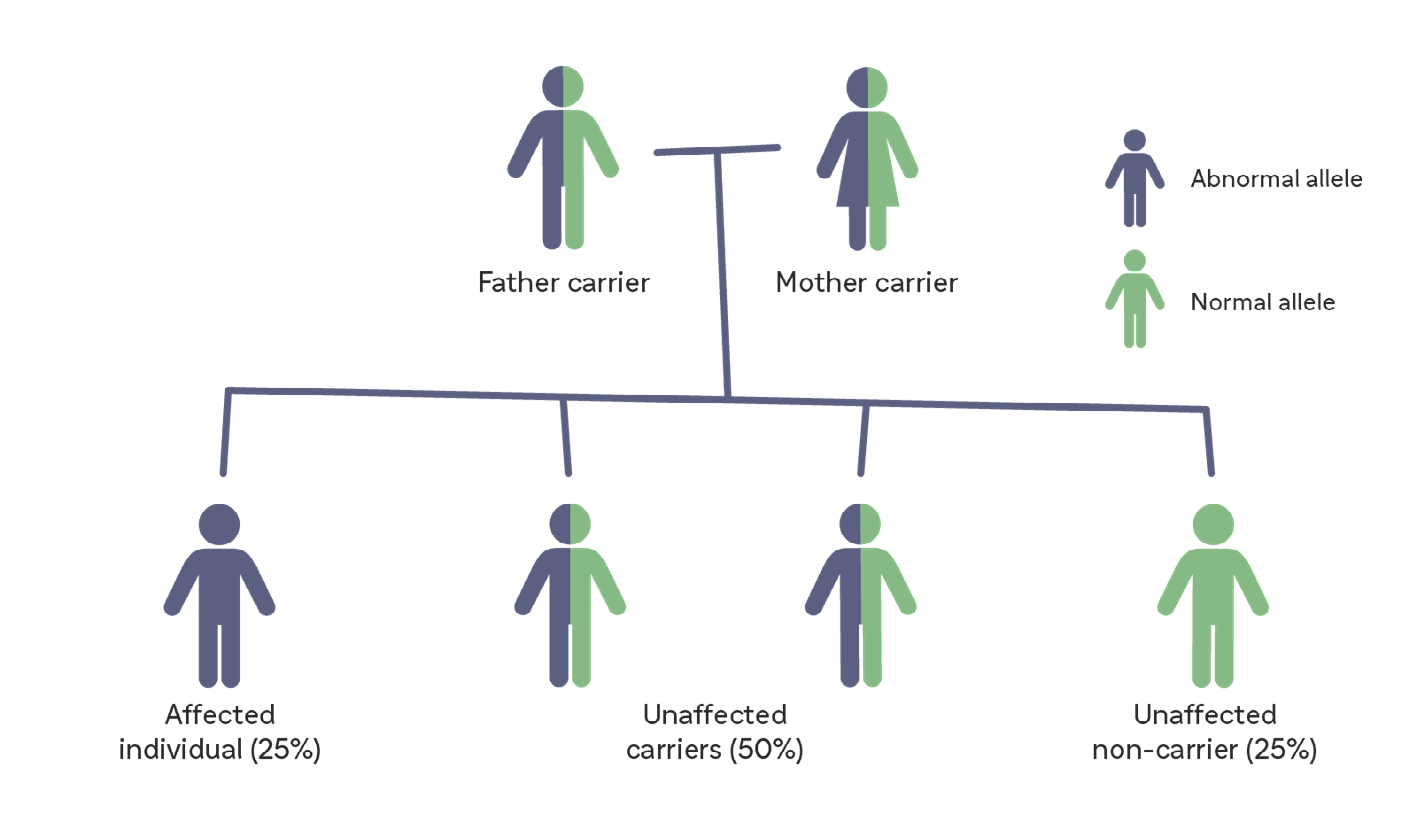
When there is a GAA activity deficiency, glycogen accumulation progressively impairs skeletal, respiratory, and smooth muscle function.2,3
Glycogen accumulation can begin early in the Pompe disease process, before the onset of detectable symptoms, and lead to extensive and irreversible muscle damage.7
This early progression in Pompe disease pathology highlights the importance of early diagnosis and access to disease-specific care.1,5,7
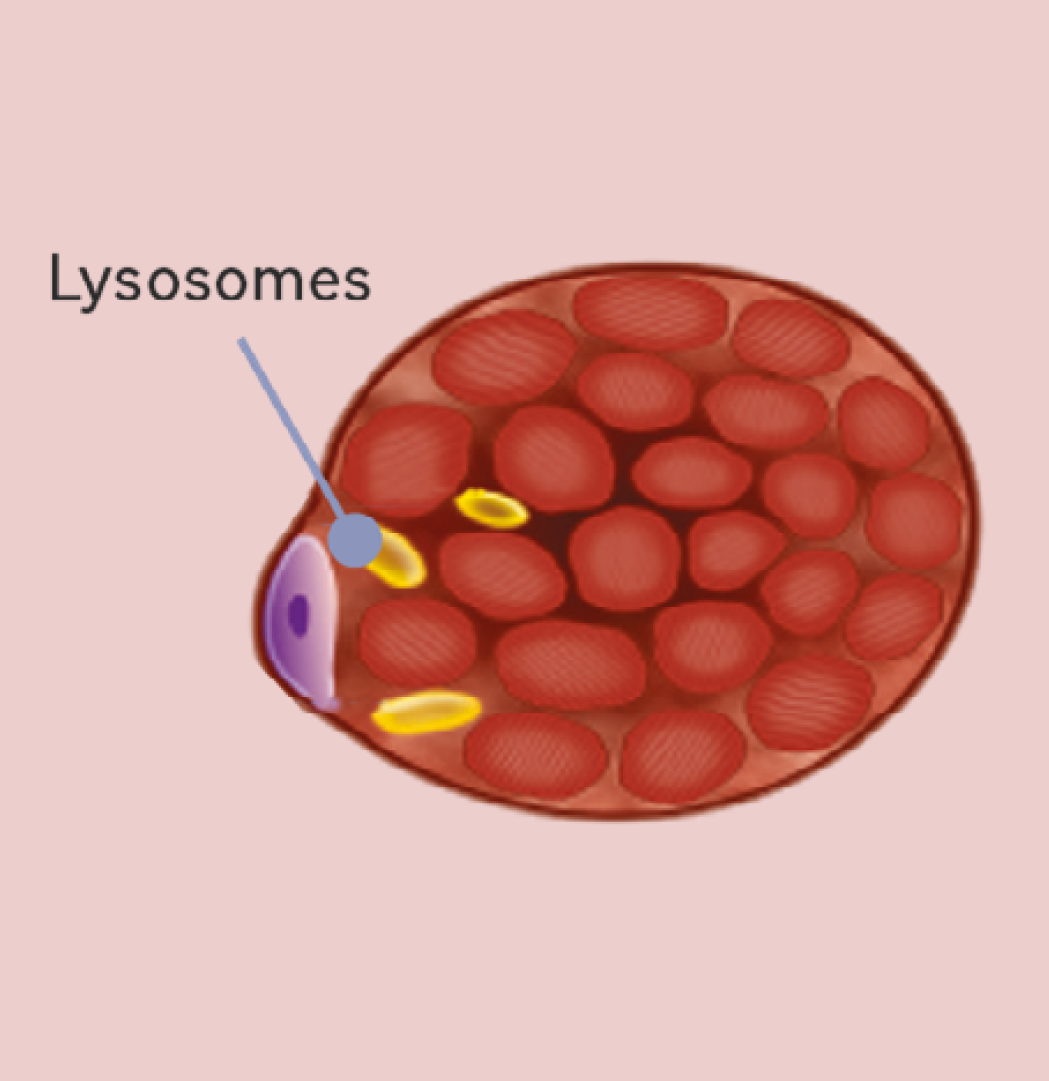
1. Pompe disease starts with a few glycogen-filled lysosomes, a stage where early muscle impairment may not be clinically detectable7
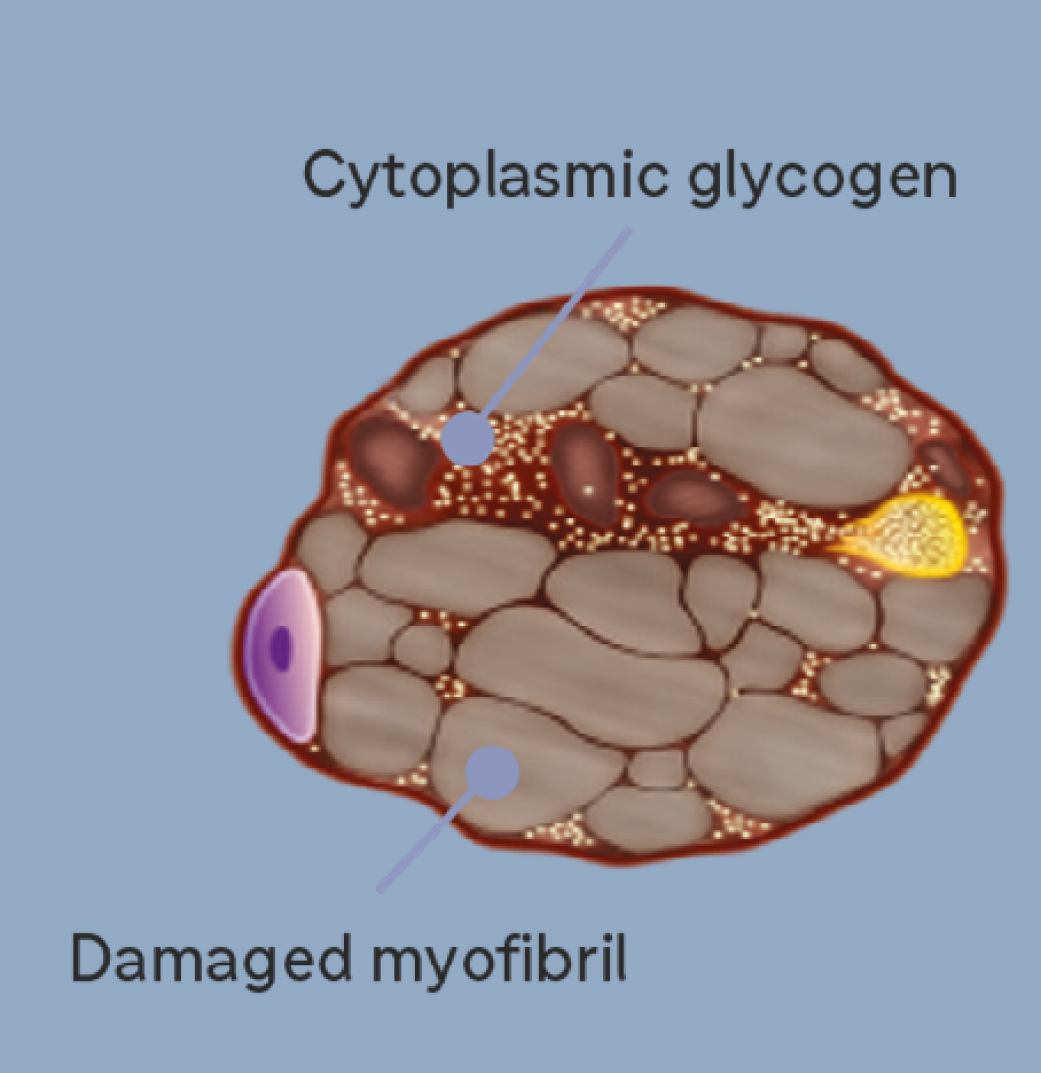
2. As glycogen accumulates, lysosomes expand and start to rupture causing progressive muscle weakness and functional impairment7
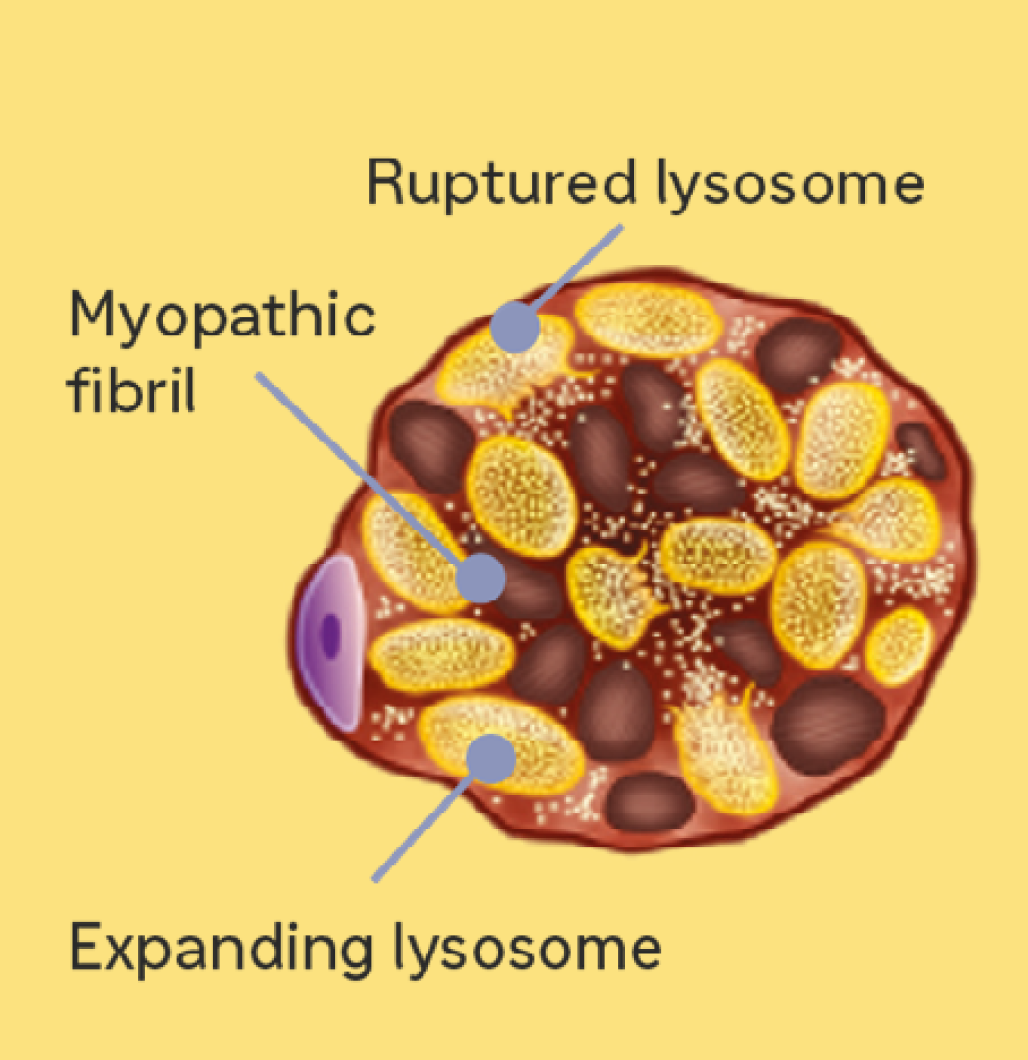
3. Increasing cytoplasmic glycogen damages the contractile structure of myofibrils, resulting in the development of more severe symptoms7
What are the different types of Pompe disease?
There are two main types of Pompe disease:
1: Late-onset Pompe disease (LOPD) and 2: Infantile-onset Pompe disease (IOPD)
LOPD |
IOPD |
| Symptoms can first present at any age3. | Onset is typically within the first few months of life 3,8 |
| Slowly progressive2,3 | Severe and rapid progression3 |
| GAA enzyme activity is usually between 1% and 40% of normal9 | GAA enzyme activity may be completely missing; if some remains, it is usually <1% of normal9 |
LOPD and IOPD differ in terms of prevalence, age of onset, level of GAA enzyme activity, severity, and progression.2,3,4,9
Although there is a crossover of clinical symptoms that are observed in both LOPD and IOPD, there are some key differences.
Cardiac symptoms are often seen early in IOPD, but are rare in people with LOPD.2,8,10
L
References
- Kishnani P et al. Am J Med Genet 2013; 161A(10): 2431–2443.
- Hirschhorn R and Reuser A. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: The Metabolic and Molecular Bases of Inherited Disease. 2014; 3389–3420.
- Kishnani P et al. Genet Med 2006; 8(5): 267–288.
- Ausems M et al. Eur J Hum Gen 1999; 7: 713–716.
- American Association of Neuromuscular & Electrodiagnostic Medicine 2009; 40(1): 149–160.
- Pompe Disease (GSD2). Association for Glycogen Storage Disease 2023. Available at: https://agsd.org.uk/all-about-gsd/ gsd-variants/pompe-disease-gsd2/. Accessed: May 2023.
- Thurberg B et al. Lab Invest 2006; 86(12): 1208–1220.
- Kishnani P et al. J Pediatr 2006; 148(5): 671–676.
- Kishnani P and Howell R. J Pediatr 2004; 144(5): S35–S43.
- Chan J et al. Mol Genet Metab 2017; 120(3): 163–172.
MAT-XU-2202602 (v5.0) Date of preparation: January 2025