
Profile

Length of experience
Myozyme® was approved in the EU in 20061

Patient groups
Myozyme® can change the natural history of Pompe disease, for patients regardless of age, gender or symptoms1,3

Early treatment initiation
Early initiation of treatment has been linked to a greater benefit and better clinical outcomes in IOPD and LOPD3,4
Evidence
Myozyme® is indicated for long-term enzyme replacement therapy (ERT) in patients with a confirmed diagnosis of Pompe disease (acid alpha glucosidase deficiency).1
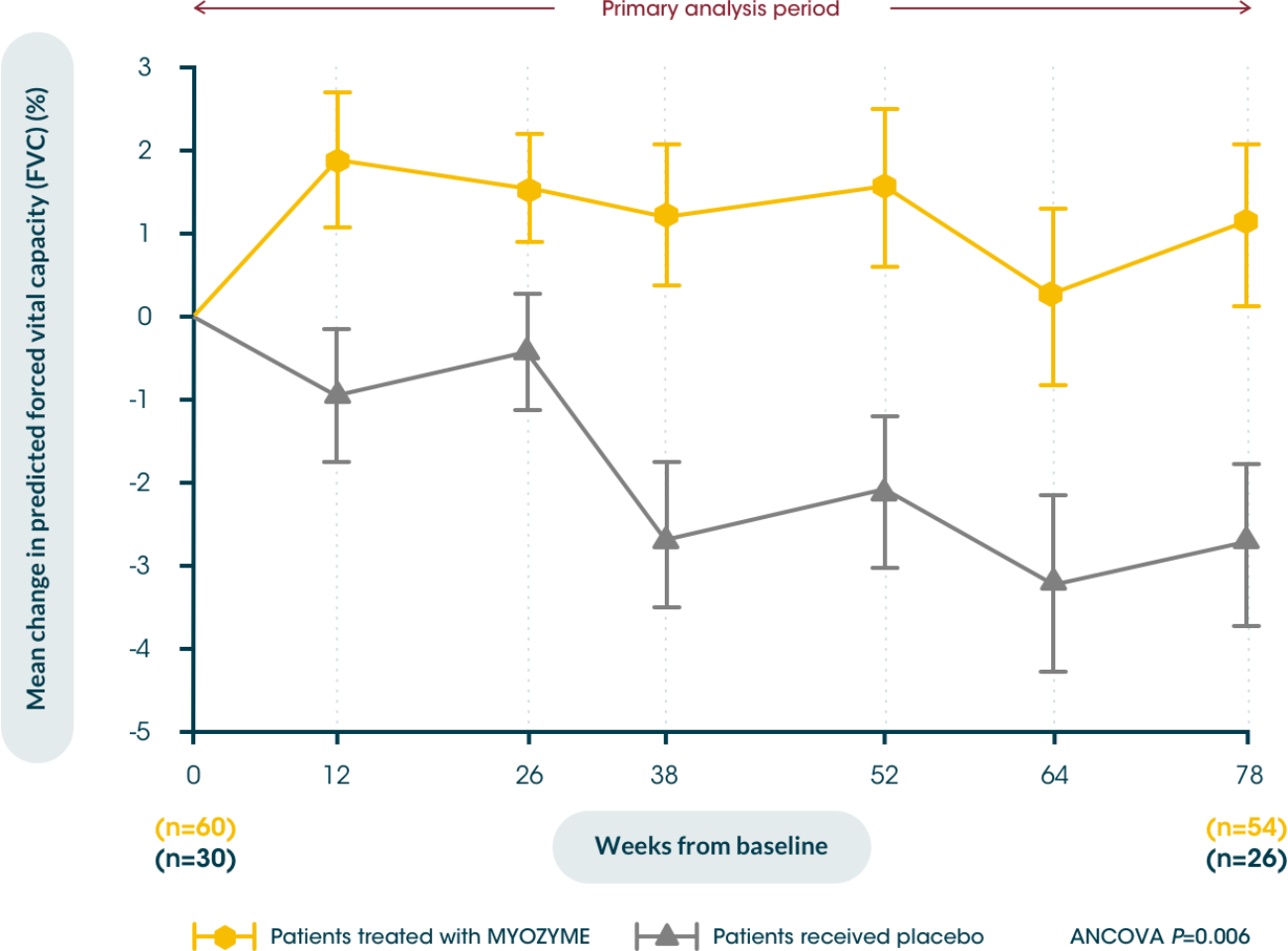
Myozyme® stabilises pulmonary function in patients with LOPD5
Patients treated with Myozyme® maintained improvements from baseline in walking distance, measured by the 6MWT, for up to 104 weeks vs. placebo5,6
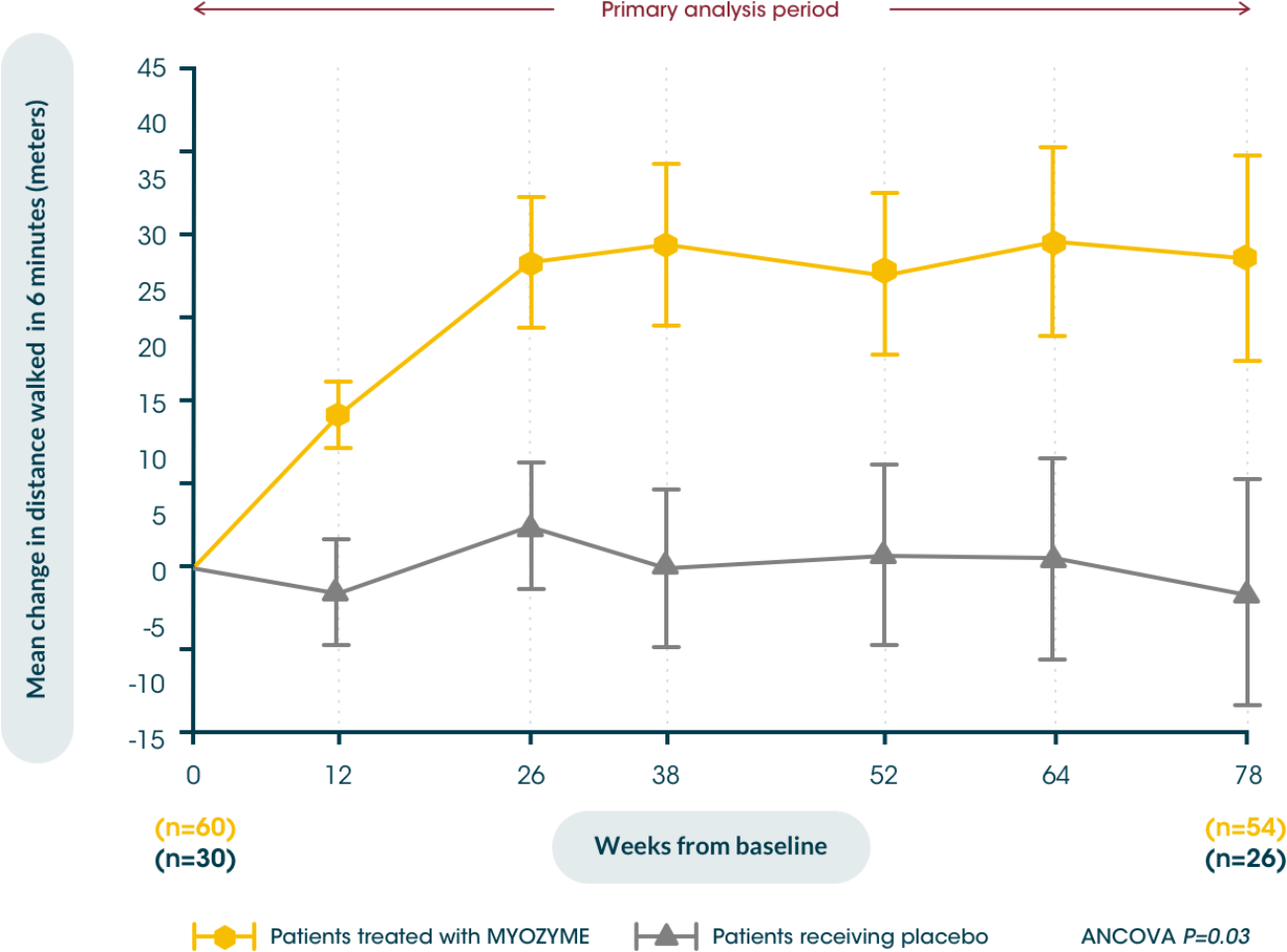
Early initiation of treatment helps improve patients’ walking distance from baseline, potentially maximising their independence vs. placebo5,6
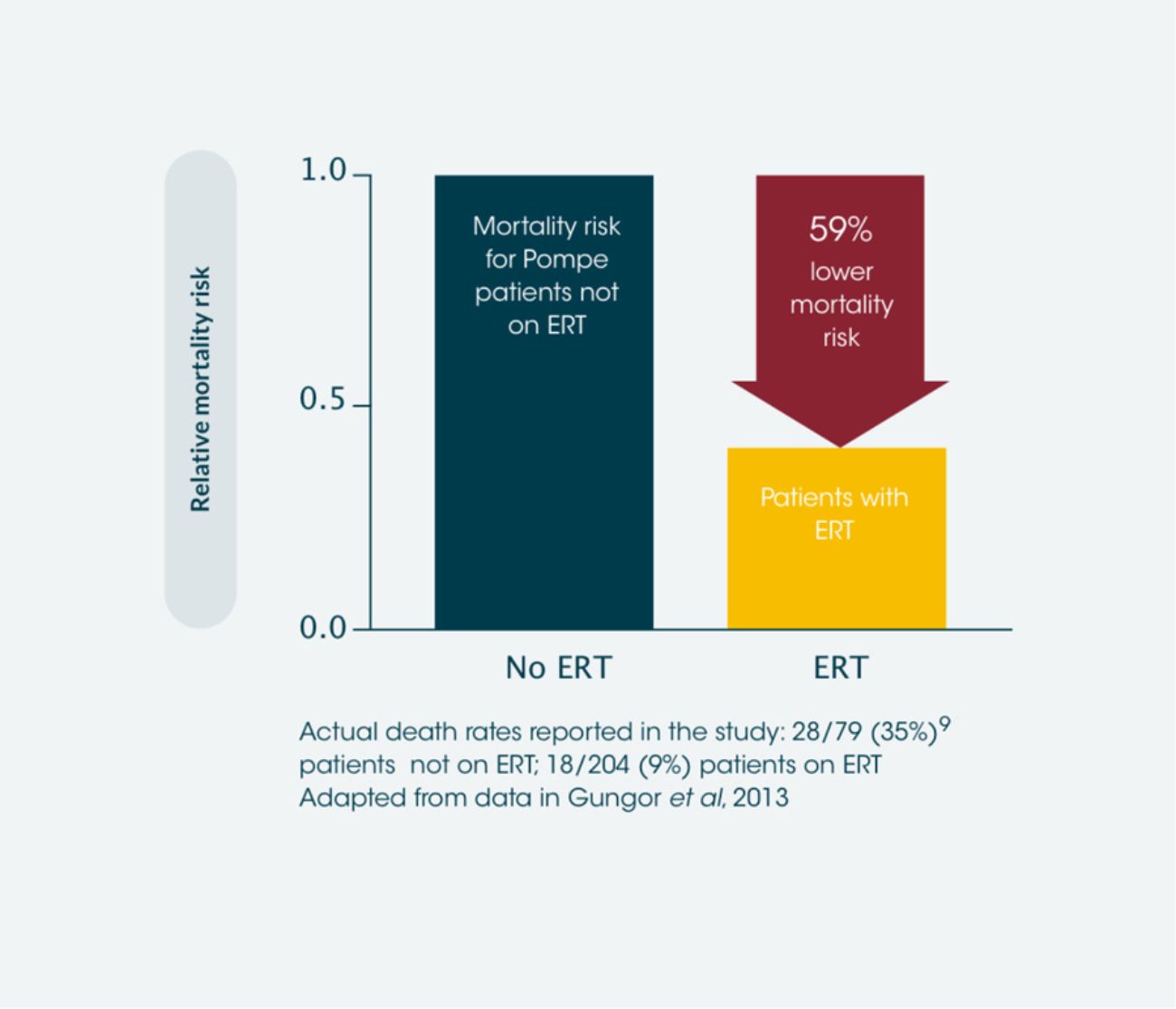
ERT results in significant improvements in survival of late-onset Pompe disease patients vs. those not treated with ERT*2
Observational data found ERT to be associated with a mortality hazard ratio of 0.41 relative to patients not receiving ERT, which is a 59% reduction in the likelihood of death. 28 patients died in the non-ERT group and 18 died in the ERT group. This extrapolates to approximately 1 extra year of life for every 8 years of ERT.**2 Use of ERT has also been shown to reduce mortality in infantile-onset Pompe disease compared to those not treated with ERT.7
*International observational study, between 2002 and 2011, including data from 283 adult patients with Pompe disease.2
ERT has a positive impact on quality of life in adult patients with Pompe disease compared to baseline.**8
**Because of the time-dependent nature of the analysis it was not possible to estimate the additional years of life gained under ERT. However, we have made ‘ ad hoc ’ calculations assuming the adjusted HR can be interpreted as a relative risk over approximately 4 years median and 8 years maximum follow-up (from start of treatment ). Using the overall raw death rate as an estimate of the raw death rate of the treated population (16 %, 46 /283), eight years of ERT would result in 1 year of life gained.
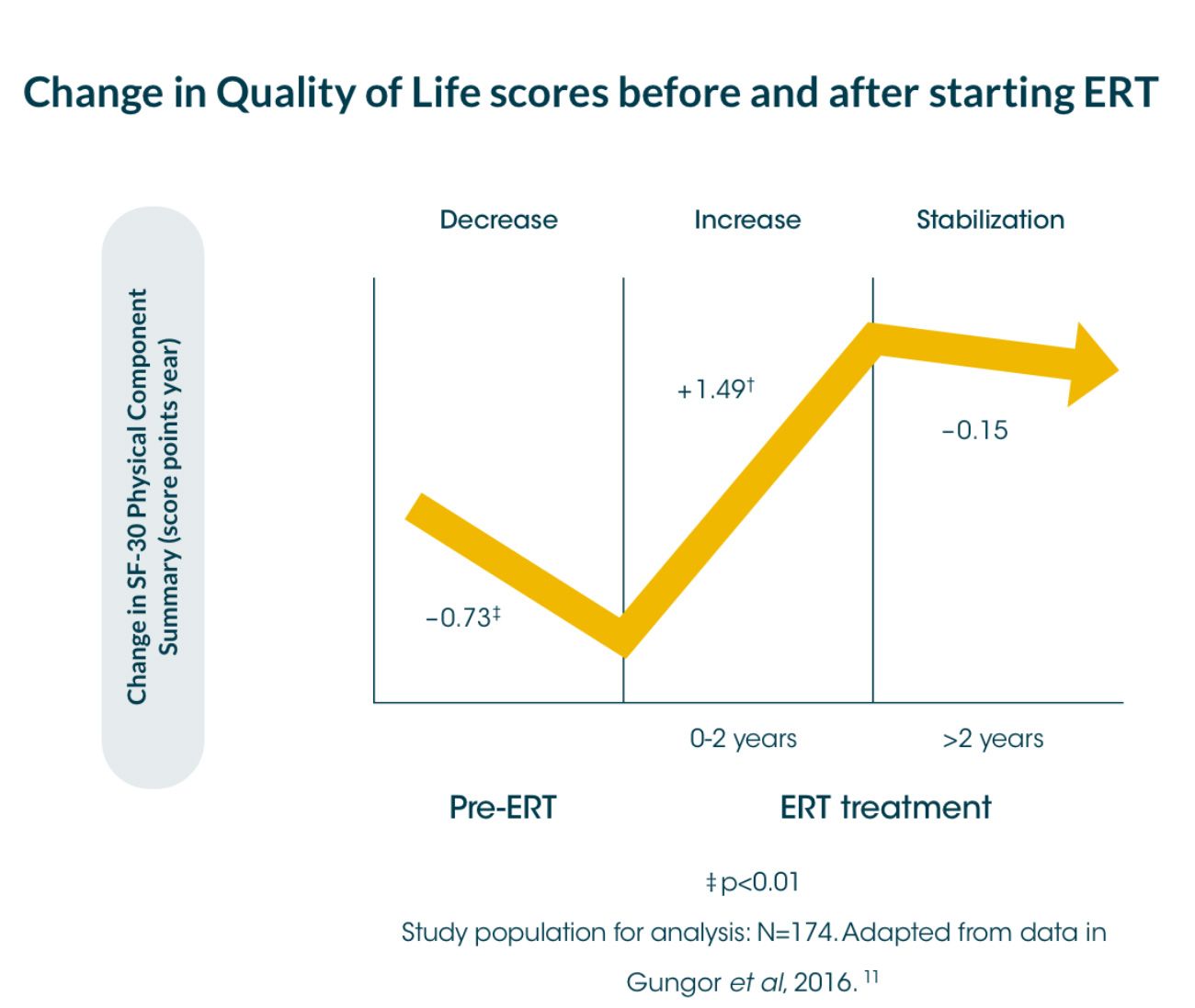
An international survey* showed that a significant decrease in the physical component summary score of the quality-of-life measure, SF-36†, in Pompe disease patients (-0.73 score points per year [sp/y]) before starting ERT.8
*International survey of 174 patients, between 2002 and 2012, assessed for quality-of-life using the SF-36 instrument, annually between 2002 and 2012; median follow-up time: 4 years (range 0.5-8)8
Myozyme® stabilises pulmonary function in patients with LOPD5
Patients treated with Myozyme® maintained improvements from baseline in walking distance, measured by the 6MWT, for up to 104 weeks vs. placebo5,6
Early initiation of treatment helps improve patients’ walking distance from baseline, potentially maximising their independence vs. placebo5,6
ERT results in significant improvements in survival of late-onset Pompe disease patients vs. those not treated with ERT*2
Observational data found ERT to be associated with a mortality hazard ratio of 0.41 relative to patients not receiving ERT, which is a 59% reduction in the likelihood of death. 28 patients died in the non-ERT group and 18 died in the ERT group. This extrapolates to approximately 1 extra year of life for every 8 years of ERT.**2 Use of ERT has also been shown to reduce mortality in infantile-onset Pompe disease compared to those not treated with ERT.7
*International observational study, between 2002 and 2011, including data from 283 adult patients with Pompe disease.2
ERT has a positive impact on quality of life in adult patients with Pompe disease compared to baseline.**8
**Because of the time-dependent nature of the analysis it was not possible to estimate the additional years of life gained under ERT. However, we have made ‘ ad hoc ’ calculations assuming the adjusted HR can be interpreted as a relative risk over approximately 4 years median and 8 years maximum follow-up (from start of treatment ). Using the overall raw death rate as an estimate of the raw death rate of the treated population (16 %, 46 /283), eight years of ERT would result in 1 year of life gained.
An international survey* showed that a significant decrease in the physical component summary score of the quality-of-life measure, SF-36†, in Pompe disease patients (-0.73 score points per year [sp/y]) before starting ERT.8
*International survey of 174 patients, between 2002 and 2012, assessed for quality-of-life using the SF-36 instrument, annually between 2002 and 2012; median follow-up time: 4 years (range 0.5-8)8
An international survey between 2002 and 2012 with responses available for 174 adult patients showed that a significant decrease in the physical component summary score of the quality-of-life measure, SF-36†, in Pompe disease patients (-0.73 score points per year [sp/y]) before starting ERT.8
During the first 2 years of ERT, the score increased significantly to 1.49 score points/year [sp/y], and stabilised thereafter (-0.15sp/y; no significant change).8 The mental component summary score was generally stable during the entire follow-up period (no significant changes from pre- to post-ERT).8 The Rotterdam Handicap Scale (RHS) stabilised after treatment with ERT (-0.02 sp/y; CI 95 % -0.17 to 0.13).8
†Short Form Health Survey-36 (SF-36) comprising eight domains: physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional and mental health8
Long-term benefits of MYOZYME treatment in LOPD
Study design9
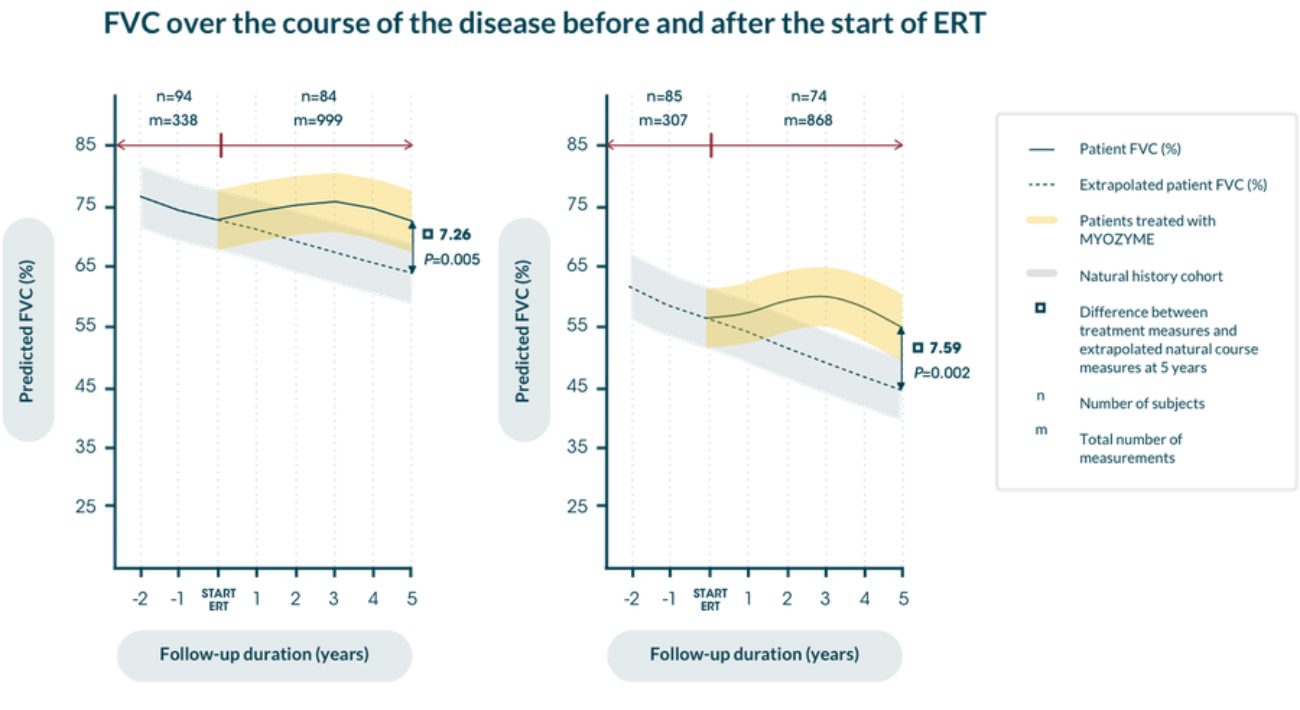
A nationwide, prospective study, open-label cohort analysed the long-term effects of enzyme replacement therapy (ERT) in 102 Dutch adult patients, ranging in age from 24 to 76 years, with Pompe disease who had not yet received ERT before enrollment on January 1, 2005. If treated, patients received Myozyme® at a dosage of 20 mg/kg every 2 weeks. Pulmonologic data and muscle function were assessed among other variables.
Clinical assessments took place every 3 to 6 months before and after the start of Myozyme; the median overall follow-up duration was 6.1 years and was compared against the natural history course extrapolated data.
Distance walked in 6MWT over the course of disease after the start of ERT9
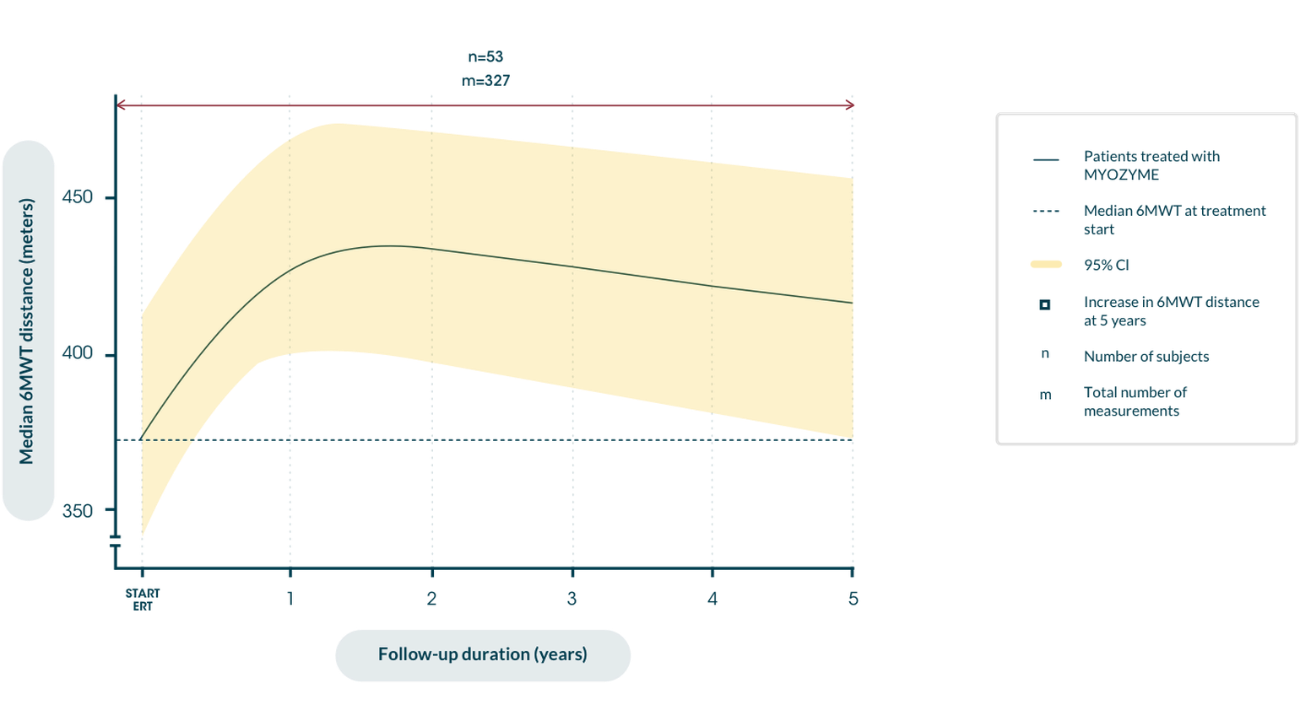
Maximizing independence in the long term9 reduces risk of wheelchair dependence10
Myozyme® can help keep long-term independence possible for adult patients with LOPD9
Individual patient response to ERT treatment on the Rasch-built Pompe-specific activity scale (R-PAct)9
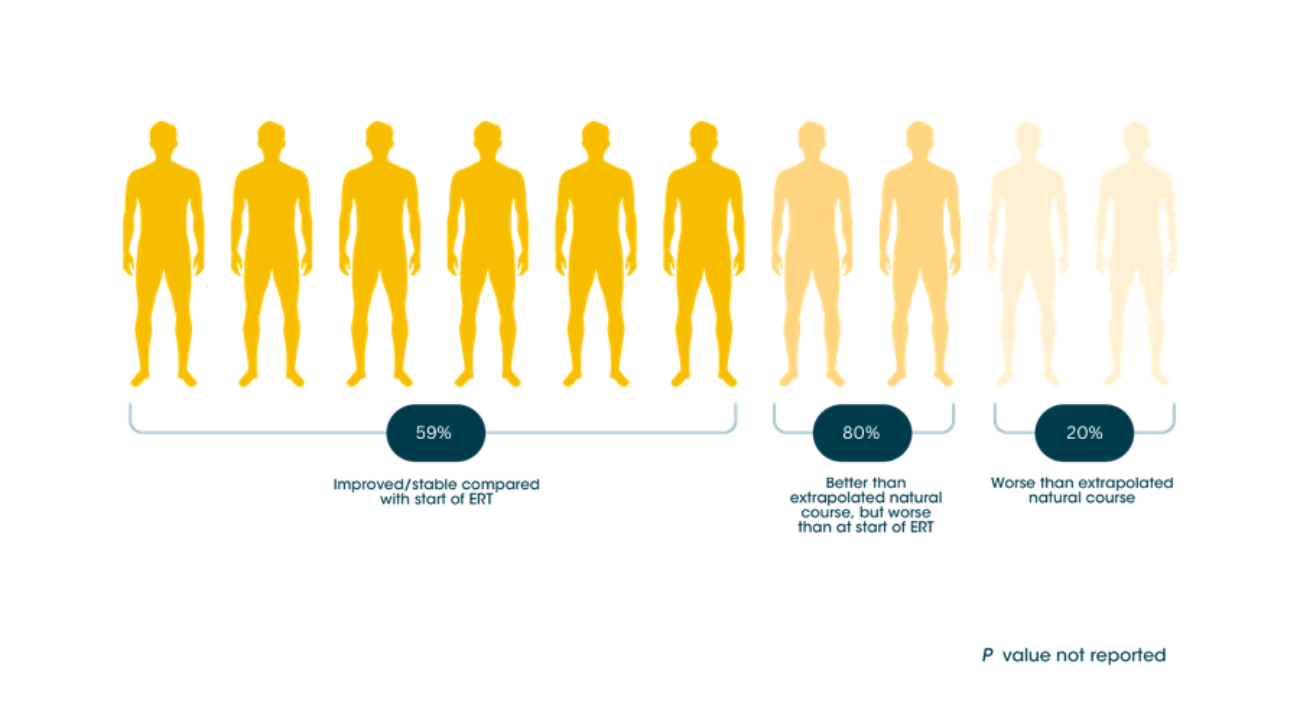
Myozyme® can help keep long-term independence possible for adult patients with LOPD9
Distance walked in 6MWT over the course of disease after the start of ERT9
Maximizing independence in the long term9 reduces risk of wheelchair dependence10
Myozyme® can help keep long-term independence possible for adult patients with LOPD9
Individual patient response to ERT treatment on the Rasch-built Pompe-specific activity scale (R-PAct)9
Myozyme® can help keep long-term independence possible for adult patients with LOPD9
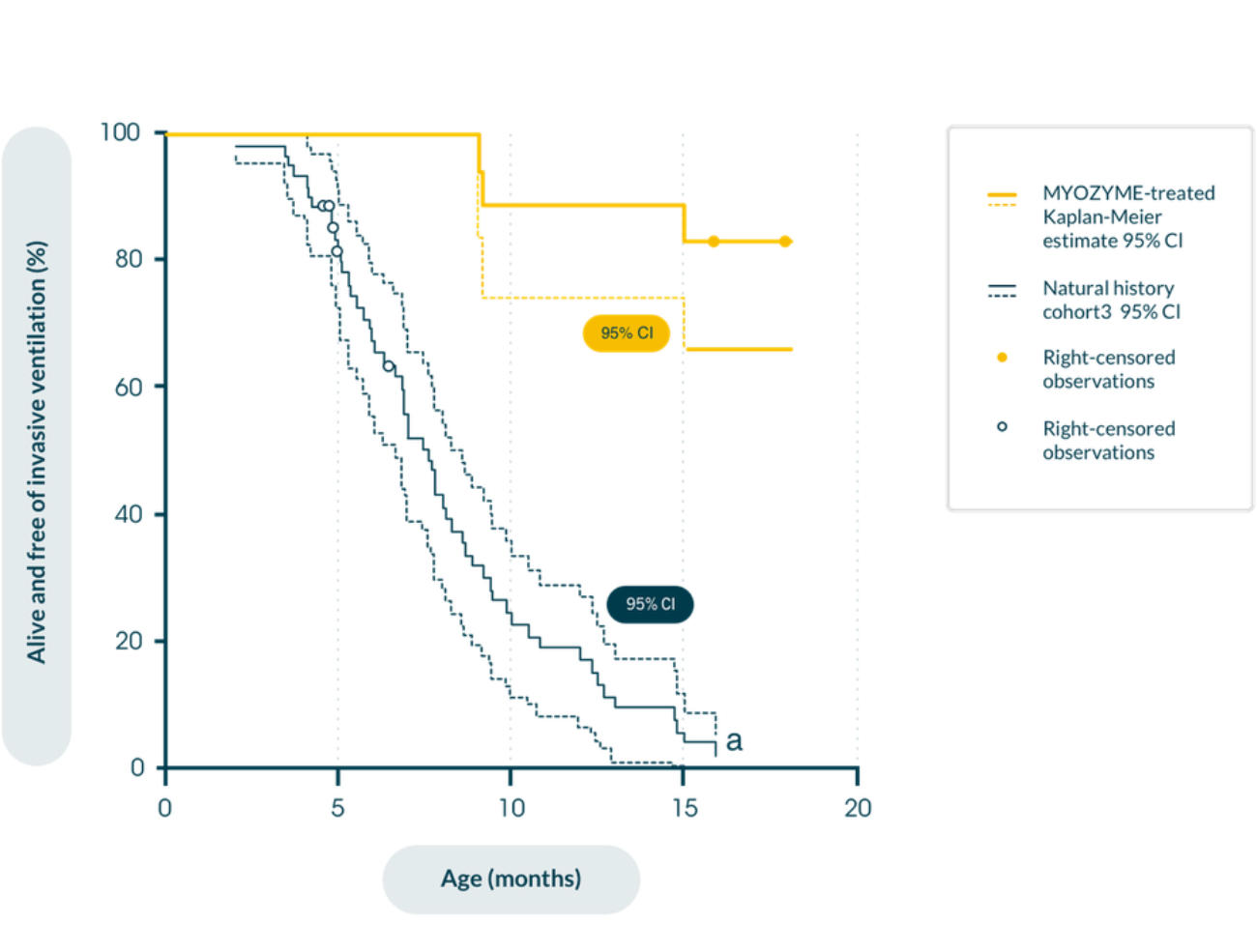
In patients with IOPD in a retrospective natural history study in patients with infantile-onset Pompe disease (n=168), early initiation of Myozyme® improves clinical outcomes in maintaining motor function and prolonging survival versus historical, untreated controls1,11
Kaplan-Meier estimate of time from birth to invasive ventilator use or death11
Summary of the safety profile
Infantile-onset Pompe disease
In clinical trials, 39 infantile-onset patients were treated with Myozyme for more than three years (168 weeks with a median of 121 weeks;). Adverse reactions were mostly mild to moderate in intensity and almost all occurred during the infusion or during the 2 hours following the infusion (infusion associated reactions, IARs). Serious infusion reactions including urticaria, rales, tachycardia, decreased oxygen saturation, bronchospasm, tachypnoea, periorbital oedema and hypertension have been reported.
Late-onset Pompe disease
In a placebo-controlled study lasting 78 weeks, 90 patients with late-onset Pompe disease, aged 10 to 70 years, were treated with Myozyme or placebo randomized in a 2:1 ratio. Overall, the numbers of patients experiencing adverse reactions and serious adverse reactions were comparable between the two groups. The most common adverse reactions observed were IARs. Slightly more patients in the Myozyme group than in the placebo group experienced IARs (28% versus 23%). The majority of these reactions were non-serious, mild to moderate in intensity and resolved spontaneously. Serious adverse reactions reported in 4 patients treated with Myozyme were: angioedema, chest discomfort, throat tightness, non-cardiac chest pain and supraventricular tachycardia. Reactions in 2 of these patients were IgE-mediated hypersensitivity reactions.1
References
- Myozyme Summary of Product Characteristics. Accessed January 2025.
- Güngör D, et al. Orphanet J Rare Dis. 2013;8:49.
- Kishnani PS, et al. Genet Med. 2006; 8(5):267–288.
- Barba-Romero MA, et al. Rev. Neurol. 2012;54(8):497–507.
- van der Ploeg AT, et al. N Engl J Med. 2010;362(15):1396-1406.
- van der Ploeg AT, et al. Mol Genet Metab. 2012;107(3):456-461.
- Chen M, et al. Cochrane Database Syst Rev. 2017;11:CD011539.
- Güngör D, et al. J Inherit Metab Dis. 2016;39(2):253–260.
- Kuperus E, et al. Neurology. 2017;89(23):2365–2373.
- van der Meijden JC, et al. Orphanet J Rare Dis. 2018;13(1):82.
- Kishnani PS, et al. Neurology. 2007;68(2):99-109.
MAT-XU-2302935 (v2.0) Date of preparation: January 2025