
THE FEATURES OF NUVAXOVID™ JN.1 ▼
a non-mRNA, protein-based COVID-19 vaccine
and older.
The use of this vaccine should be in accordance with official recommendations
| Proven Efficacy | Safety Profile | Fridge Stable Storage |
|---|---|---|
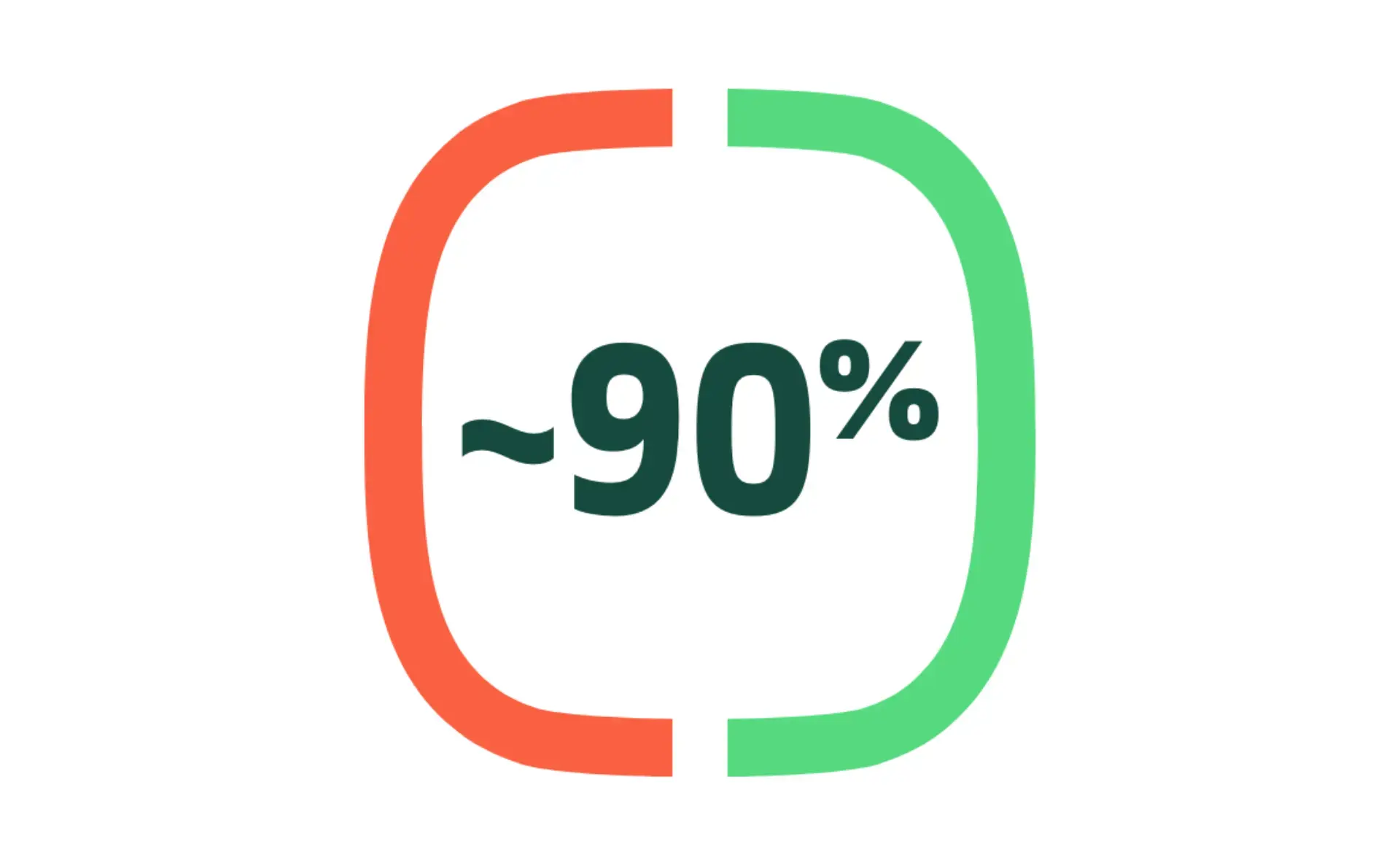
 NUVAXOVID has demonstrated ~90% (0.69% absolute risk reduction) effective against COVID-19 in Phase 3 clinical trials vs placebo1,2*
|
 Nuvaxovid helped protect patients from COVID-19, and was generally well tolerated in clinical trials1
|
 Store in a refrigerator and administration with prefilled syringes1
|
* In a Phase 3, randomised, observer-blinded, placebo-controlled PREVENT 19 trial in ≥18 years; Nuvaxovid demonstrated a vaccine efficacy of 90.4% (0.69% absolute risk reduction)(95% CI: 82.9–94.6%) against PCR-confirmed symptomatic COVID-19 vs placebo, assessed from 7 days after the second dose. The efficacy of NUVAXOVID JN.1 is inferred from the efficacy data of the NUVAXOVID (Original, Wuhan strain) vaccine and immunogenicity data from the adapted vaccine of the Omicron BA.5 strain.1 Before administration, please see the full SmPC for NUVAXOVID JN.1. | ||
Efficacy in preventing COVID-19 vs placebo in PREVENT-19 (US/Mexico) and 2019nCoV-302 Study
(UK study)1-3
PREVENT-19 STUDY: NUVAXOVID was 90.4% effective in preventing COVID-19 in this pivotal study of almost 25,000 patients (0.69% ARR)(95% CI: 82.9, 94.6)*
UK STUDY: NUVAXOVID was 89.7% effective in preventing COVID-19 in this pivotal study of over 14,000 patients (1.57% ARR)(95% CI: 80.2, 94.6)†
|
Efficacy in preventing COVID-19
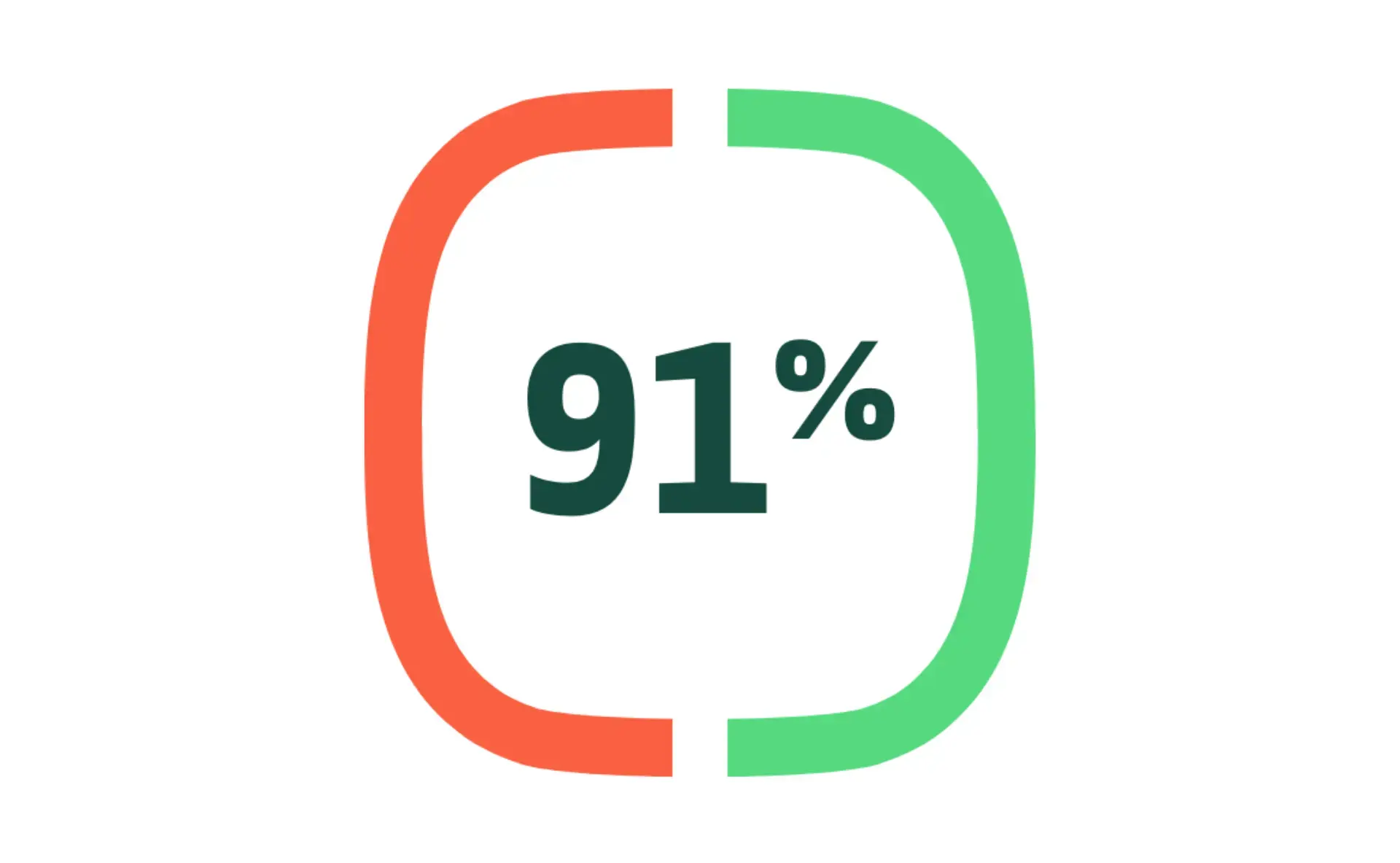
vs placebo in patient with coexisting illness in PREVENT-19 and 2019nCoV-302 Study (UK study)1-3 (sub group analysis) PREVENT-19 STUDY: NUVAXOVID was 91.0% effective in preventing COVID-19 in patients with coexisting illness (0.72% ARR)(95% CI: 83.6, 95.0)*
UK STUDY: NUVAXOVID was 90.9% effective in preventing COVID-19 in patients with coexisting illness (0.95% ARR)(95% CI: 70.4, 97.2)†
|
* Phase 3, multicenter, randomized, observer-blinded, placebo-controlled clinical trial evaluating efficacy and safety of 29,582 total participants aged 18 years and older. 25,452 participants were included in the per-protocol efficacy analysis population. Primary endpoint was efficacy in preventing PCR-confirmed symptomatic mild, moderate, or severe COVID-19 from 7 days after the second dose 90.4% (95% CI: 82.9, 94.6), p<0.001, N=25,452. The primary endpoint was met. The assessment of coexisting conditions is based on the Centers for Disease Control and Prevention definitions of persons at risk for complications of Covid-19. Participants at overall high risk for Covid-19 included those 65 years of age or older and those of any age with chronic health conditions or an increased risk for Covid-19 because of work or living conditions.
† Phase 3, multicenter, randomized, observer-blinded, placebo-controlled study conducted in a total of 15,187 participants aged 18-84. 14,039 were included in the per-protocol efficacy analysis population. Primary endpoint was virologically confirmed mild, moderate, or severe COVID-19 infection with an onset at least 7 days after the second injection in patients who were serologically negative at baseline 89.7% (95% CI: 80.2, 94.6). The primary endpoint was met. Participants had at least one coexisting condition that had been defined by the Centers for Disease Control Prevention as a risk factor for severe Covid-19. These conditions included chronic respiratory, cardiac, renal, neurologic, hepatic, and immune- noncompromising conditions as well as obesity.
The efficacy of NUVAXOVID JN.1 is inferred from the efficacy data of the NUVAXOVID (Original, Wuhan strain) vaccine and immunogenicity data from the adapted vaccine of the Omicron BA.5 strain.1
Before administration, please see the full SmPC for NUVAXOVID JN.1.
Safety Profile
| ADULTS AGED 18+ | ADOLESCENTS AGED 12-17 | ||
The safety of NUVAXOVID was evaluated across 5 clinical trials with a total of 49,950 participants aged 18 years and older received at least one dose of the two-dose primary series of Nuvaxovid (n=30,058) or placebo (n=19,892).1 | The safety of NUVAXOVID was evaluated in an interim analysis of the paediatric expansion portion of an ongoing Phase 3 multicentre, randomised, observer-blinded, placebo-controlled study. Safety data were collected in 2,232 patients aged 12 through 17 years who received at least one dose of NUVAXOVID or placebo.1 | ||
 | ≈50,000 were evaluated across 5 clinical trials1 |  | Median duration of adverse reactions was ≤1 day for systemic events and ≤2 days for local events1 |
 | Adverse reactions were usually mild to moderate in severity1-3 | | Adverse reactions were usually mild to moderate severity1 |
 | Patients aged 65+ experienced a lower incidence of adverse reactions than patients aged 18-641 | ||
Participants 18 years of age and older
The safety of Nuvaxovid was evaluated from an interim analysis of pooled data from 5 ongoing clinical trials conducted in Australia, South Africa, the United Kingdom, the United States and Mexico. At the time of the analysis, a total of 49,950 participants aged 18 years and older received at least one dose of the two-dose primary series of Nuvaxovid (n=30,058) or placebo (n=19,892). At the time of vaccination, the median age was 48 years (range 18 to 95 years). The median duration of follow-up was 70 days post-Dose 2, with 32,993 (66%) participants completing more than 2 months follow-up post-Dose 2.
Of the pooled reactogenicity data, which includes participants aged 18 years and older enrolled in the two phase 3 studies who received any dose of Nuvaxovid (n=20,055) or placebo (n=10,561), the most frequent adverse reactions were injection site tenderness (75%), injection site pain (62%), fatigue (53%), myalgia (51%), headache (50%), malaise (41%), arthralgia (24%), and nausea or vomiting (15%). Adverse reactions were usually mild to moderate in severity with a median duration of less than or equal to 2 days for local events and less than or equal to 1 day for systemic events following vaccination.
Overall, there was a higher incidence of adverse reactions in younger age groups: the incidence of injection site tenderness, injection site pain, fatigue, myalgia, headache, malaise, arthralgia, and nausea or vomiting was higher in adults aged 18 to less than 65 years than in those aged 65 years and above.
Local and systemic adverse reactions were more frequently reported after Dose 2 than after Dose 1.
Licensed inactivated seasonal influenza vaccines were co-administered to participants on the same day as Dose 1 of Nuvaxovid (n=217) or placebo (n=214) in the opposite deltoid muscle of the arm in 431 participants enrolled in an exploratory Phase 3 (2019nCoV-302) sub-study. The frequency of local and systemic adverse reactions in the influenza sub-study population was higher than in the main study population following Dose 1 in both Nuvaxovid and placebo recipients.
Adolescents 12 through 17 years of age
The safety of Nuvaxovid in adolescents was evaluated in an interim analysis of the paediatric expansion portion of an ongoing Phase 3 multicentre, randomised, observer-blinded, placebo-controlled study (Study 2019nCoV-301). Safety data were collected in 2,232 participants 12 through 17 years of age, with and without evidence of prior SARS-CoV-2 infection, in United States who received at least one dose of Nuvaxovid (n=1,487) or placebo (n=745). Demographic characteristics were similar among participants who received Nuvaxovid and those who received placebo.
The most frequent adverse reactions were injection site tenderness (71%), injection site pain (67%), headache (63%), myalgia (57%), fatigue (54%), malaise (43%), nausea or vomiting (23%), arthralgia (19%) and pyrexia (17%). Fever was observed more frequently in adolescents aged 12 through to 17 years compared to adults, with the frequency being very common after the second dose in adolescents. Adverse reactions were usually mild to moderate in severity with a median duration of less than or equal to 2 days for local events and less than or equal to 1 day for systemic events following vaccination.
Participants 18 years of age and older
In an independent study (CoV-BOOST study, EudraCT 2021-002175-19) evaluating the use of a Nuvaxovid booster dose in individuals who had completed primary vaccination with an authorised mRNA COVID-19 vaccine or adenoviral vector COVID-19 vaccine, no new safety concerns were identified.
The safety and immunogenicity of a booster dose of Nuvaxovid was evaluated in an ongoing Phase 3, multicenter, randomized, observer-blinded, placebo-controlled study (Study 2019nCoV-301). Overall, 12,777 participants received a booster dose of the vaccine at least 6 months after the two-dose primary series (median of 11 months between completion of primary series and booster dose). Of the 12,777 participants who received a booster dose, 39 participants did not receive Nuvaxovid for all three doses. The safety analyses included evaluation of solicited local and systemic adverse reactions within 7 days after a booster dose for participants who completed the electronic diary (n=10,137).
The most frequent solicited adverse reactions were injection site tenderness (73%), injection site pain (61%), fatigue (52%), muscle pain (51%), headache (45%), malaise (40%), and joint pain (26%).
Adolescents 12 through 17 years of age
The safety of a booster dose of Nuvaxovid was evaluated in an interim analysis of an ongoing Phase 3 study (Study 2019nCoV-301). A total of 1,499 participants received a booster dose approximately 9 months after receiving Dose 2 of the primary series. A subset of 220 participants who received the booster dose were evaluated for solicited adverse reactions within 7 days after the booster dose (Ad Hoc Booster Safety Analysis Set), of whom 190 completed the electronic diary.
Solicited adverse reactions occurred at higher frequencies and with higher grade in adolescents compared to adults. The most frequent solicited adverse reactions were injection site tenderness (72%), headache (68%), fatigue (66%), injection site pain (64%), muscle pain (62%), malaise (47%), and nausea/vomiting (26%) with a median duration of 1 to 2 days following vaccination. No new safety concerns from the time of the booster dose administration through 28 days after administration were noted among participants.
For a full list of adverse events please refer to the Nuvaxovid JN.1 SmPC.
|
NUVAXOVID showed similar reactogenicity profile to control group (MenACWY) in a UK blinded, randomised Phase 2 trial4
|

A multicentre, blinded, randomised, Phase 2 clinical study aimed to assess the safety, reactogenicity, and immunogenicity of homologous and heterologous COVID-19 boosters in ≥30-year-old UK adults. The study was not powered to make comparisons between vaccines, but all vaccine arms were compared to the active control, MenACWY (Pfizer, Inc.) vaccine, separated into groups A, B, and C. Participants aged 30 years and older were randomly assigned to one of three study groups with equal probability within groups A–C. Group A received NVX-CoV2373 (protein-based, Novavax, Inc., 5μg rS + 50μg Matrix-M™), or MenACWY. Group B received BNT162b2 (mRNA, Pfizer, Inc., 30μg) or MenACWY. Group C received mRNA-1273 (mRNA, Moderna, Inc., 100μg, twice the currently approved dosage) or MenACWY. The occurrence of solicited reactogenicity events following the third vaccination was recorded in participant electronic diaries were recorded daily for 7 days. The percentage of participants who reported local events (pain, warmth, redness, itch, swelling, and/or hardness) and systemic events (malaise, muscle ache, fatigue, headache, joint pain, fever, feverishness, diarrhoea, and/or nausea) after vaccination were measured. Feverishness was defined as feeling unwell/shivery but not always associated with measurable fever.
Limitations
- The population included a primarily seronegative cohort of mostly White people ≥30 years of age.
- The grouped study design did not permit randomisation of all vaccines simultaneously. Instead, each COVID-19 vaccine arm included a MenACWY active control group. Hence, the frequency of the reported reactogenicity symptoms might vary partially due to population differences in different groups.
- The dose concentration of the mRNA-1273 vaccine used at the time of this study (100μg) was twice the subsequently approved dose concentration for adults receiving their third vaccine dose.
- The trial was not powered to directly compare reactogenicity.
- Study participants had no history of laboratory-confirmed SARS-CoV-2 infection.
- The incidence of symptoms among the different age groups was not stratified in the present analysis.
|
NUVAXOVID JN.1 can be stored in a refrigerator and is administered with prefilled syringes1
|
| NUVAXOVID JN.11 | |
| Injection type | Pre-filled syringe (0.5mL) |
| Shelf life | 9 months at 2-8°C |
| Pack size | 10 pre-filled syringes |
Store prefilled syringes between 2°C to 8°C(36°F to 46°F).
- NUVAXOVID JN.1 [Summary of Product Characteristics]. Sanofi; 2025
- Dunkle LM, Kotloff KL, Gay CL, et al; 2019nCoV-301 Study Group. Efficacy and safety of NVX-CoV2373 in adults in the United States and Mexico. N Engl J Med. 2022;386(6):531-543. doi:10.1056/NEJMoa2116185
- Heath PT, Galiza EP, Baxter DN, et al. Safety and efficacy of NVX-CoV2373 Covid-19 vaccine. N Engl J Med. 2021;385(13):1172-1183. doi:10.1056/NEJMoa2107659
- Marchese AM, et al. Vaccine. 2025 Jan 12;44:126569. doi: 10.1016/j.vaccine.2024.126569.
MAT-XU-2600435 (v2.0) Date of Preparation April 2026