
ICARIA-MM: The first positive Phase 3 trial of an anti-CD38 monoclonal antibody triplet in relapsed and refractory multiple myeloma (RRMM).1 For adult patients with relapsed and refractory multiple myeloma improve outcome with SARCLISA® in combination with pomalidomide + dexamethasone (Pd) vs. Pd.*1,2
*SARCLISA® + Pd significantly increased mPFS vs. Pd alone (11.53 vs. 6.47 months, HR 0.596 [95% CI 0.44, 0.81]; p=0.001).1
Indication
SARCLISA® is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adult patients with relapsed and refractory multiple myeloma (MM) who have received at least two prior therapies including lenalidomide and a proteasome inhibitor (PI) and have demonstrated disease progression on the last therapy.

Efficacy
SARCLISA® + Pd (ICARIA-MM): Trial design
ICARIA-MM: The first positive Phase 3 trial of an anti-CD38 mAb-Pd triplet in RRMM.1
Studied in over 300 adult patients with relapsed and refractory MM in a Phase 3, multicentre, multinational, randomised, open-label, 2‑arm study.1
.2023-07-19-08-42-06.png)
Adapted from Attal M et al. 2019.1 *PFS results were assessed by an IRC, based on central laboratory data for M-protein, and central radiologic imaging review using the IMWG criteria.1 Median time to follow-up: 11.6 months.1
†sCR, CR, VGPR, and PR were evaluated by the IRC using the IMWG response criteria.1
‡1 patient in the SARCLISA®-Pd group was previously treated with daratumumab.1
SARCLISA® + Pd was studied in a diverse patient population.1,2
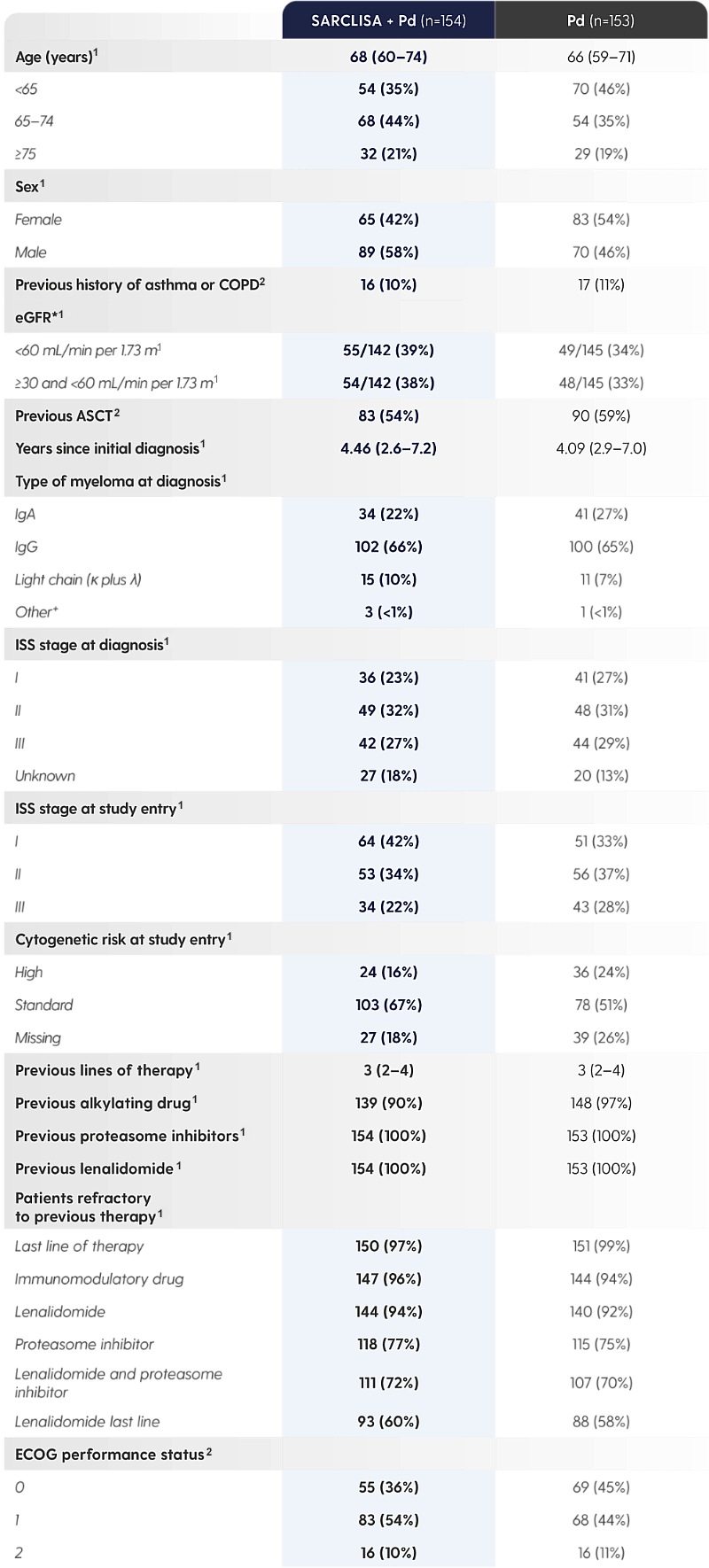
Adapted from Attal M et al. 2019 and from SARCLISA® Summary of Product Characteristics.1,2
*Information on race could not be collected in some countries, hence not all values were available.1
†IgM unknown or undetected.1
‡Central laboratory cut-off 50% for del(17p), 30% for t(4;14) and t(14;16).1
|
The ICARIA-MM patient population was heavily pre-treated and also included patients with high cytogenetic risk,* patients with renal impairment,† and patients who were elderly‡ *Central laboratory cut-off 50% for del(17p), 30% for t(4;14) and t(14;16).1 |
Studied in over 300 adult patients with relapsed and refractory MM in a Phase 3, multicentre, multinational, randomised, open-label, 2‑arm study.1
Adapted from Attal M et al. 2019.1 *PFS results were assessed by an IRC, based on central laboratory data for M-protein, and central radiologic imaging review using the IMWG criteria.1 Median time to follow-up: 11.6 months.1
†sCR, CR, VGPR, and PR were evaluated by the IRC using the IMWG response criteria.1
‡1 patient in the SARCLISA®-Pd group was previously treated with daratumumab.1
SARCLISA® + Pd was studied in a diverse patient population.1,2
Adapted from Attal M et al. 2019 and from SARCLISA® Summary of Product Characteristics.1,2
*Information on race could not be collected in some countries, hence not all values were available.1
†IgM unknown or undetected.1
‡Central laboratory cut-off 50% for del(17p), 30% for t(4;14) and t(14;16).1
|
The ICARIA-MM patient population was heavily pre-treated and also included patients with high cytogenetic risk,* patients with renal impairment,† and patients who were elderly‡ *Central laboratory cut-off 50% for del(17p), 30% for t(4;14) and t(14;16).1 |
SARCLISA® + Pd (ICARIA-MM): Trial results
SARCLISA® + Pd extended median progression free survival by 5 months vs. Pd alone.1
SARCLISA® + Pd extended mPFS by 5 months vs. Pd alone.1
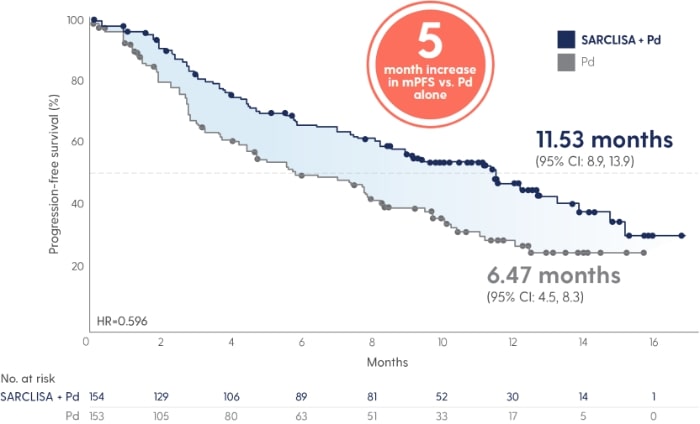
Adapted from Attal M et al. 2019.
|
40% reduction in risk of progression or death in patients receiving SARCLISA® + Pd vs. Pd alone (11.53 vs. 6.47 months [absolute change 5 months]; HR 0.596 [95% CI: 0.44, 0.81; p=0.001])1 With a median duration of follow-up of 11.6 months, median OS was not reached for either treatment group at the time of this planned interim analysis. The HR for OS was 0.69 (95% CI: 0.46, 1.02; p=0.0631).1 |
Adapted from Attal M et al. 2019.1
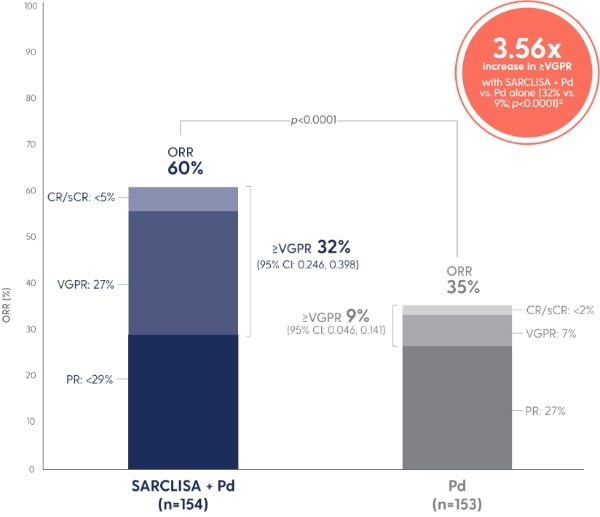
Adapted from Attal M et al. 2019.1
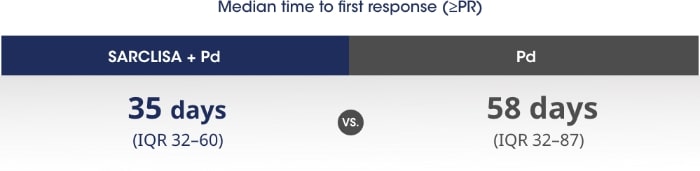
Patients responded more quickly with SARCLISA® + Pd vs. Pd alone.1
PR - partial response
A PFS benefit vs. Pd alone that was consistent across pre‑specified patient subgroups.
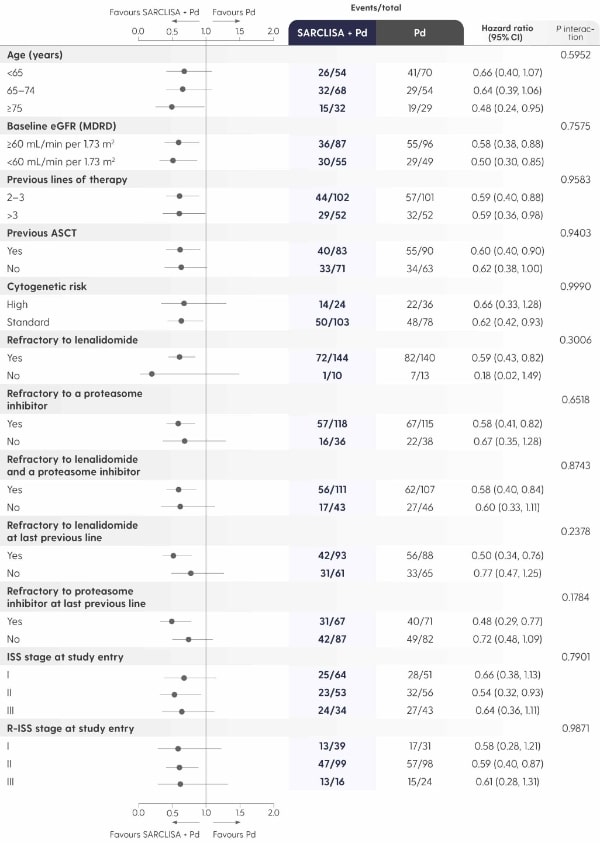
Adapted from Attal M et al. 2019.1
Safety
SARCLISA® + Pd (ICARIA-MM): Adverse Reactions
ICARIA-MM: Adverse reactions reported in patients treated with SARCLISA® + Pd vs. Pd alone.1
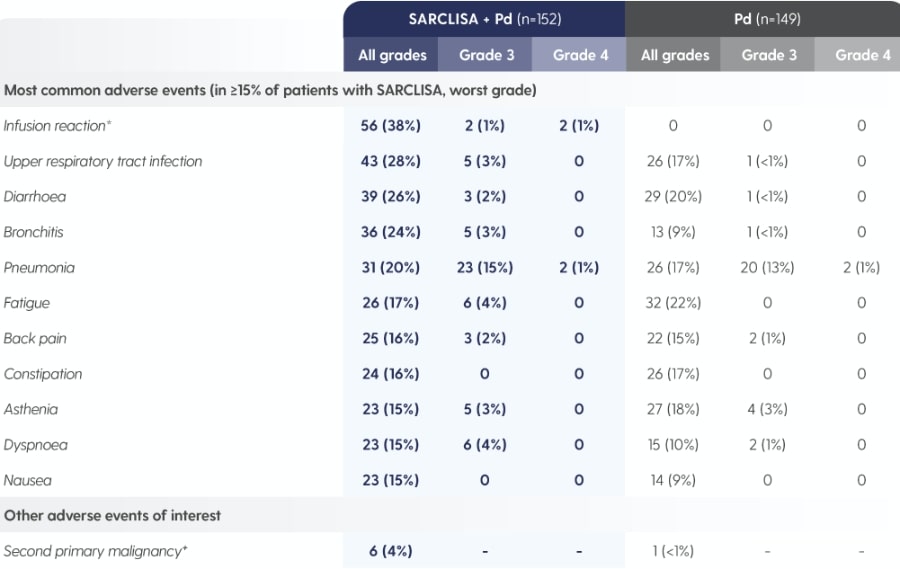
Adapted from Attal M et al. 2019.1
*As reported by the investigator in the specific form of an adverse event, including infusion-related reaction, cytokine release syndrome and drug hypersensitivity.1
†SARCLISA® + Pd group: one patient with myelodysplastic syndrome, one patient with post-radiation angiosarcoma and four patients with squamous cell carcinoma of the skin.2 Pd group: one patient with squamous cell carcinoma of the skin.2 All patients continued treatment after complete surgical excision except the patient with myelodysplastic syndrome.1
SARCLISA® + Pd (ICARIA-MM): Haematological Abnormalities
ICARIA-MM: Haematological laboratory abnormalities in patients receiving SARCLISA® + Pd vs. Pd alone.2
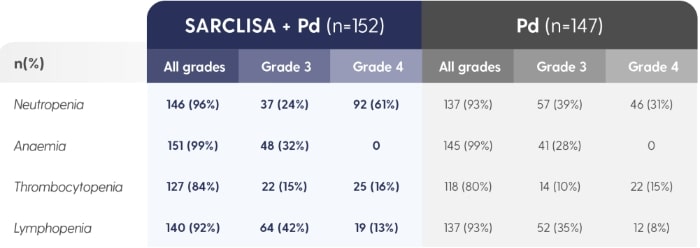
The denominator used for the percentage calculation is the number of patients with at least 1 evaluation of the laboratory test during the considered observation period.1Adapted from SARCLISA® Summary of Product Characteristics.2
30.3% patients treated with SARCLISA® had neutropenic complications.2
- Patients with neutropenia should be monitored for signs of infection
- No dose reductions of SARCLISA® are recommended
- SARCLISA® dose delays and the use of colony-stimulating factors (e.g. G-CSF) should be considered to mitigate the risk of neutropenia
Full blood cell counts should be monitored periodically during treatment.2
Overall discontinuation rate did not increase with addition of SARCLISA® to Pd4
SARCLISA® + Pd (ICARIA-MM): Adverse Reactions
ICARIA-MM: Adverse reactions reported in patients treated with SARCLISA® + Pd vs. Pd alone.1
Adapted from Attal M et al. 2019.1
*As reported by the investigator in the specific form of an adverse event, including infusion-related reaction, cytokine release syndrome and drug hypersensitivity.1
†SARCLISA® + Pd group: one patient with myelodysplastic syndrome, one patient with post-radiation angiosarcoma and four patients with squamous cell carcinoma of the skin.2 Pd group: one patient with squamous cell carcinoma of the skin.2 All patients continued treatment after complete surgical excision except the patient with myelodysplastic syndrome.1
SARCLISA® + Pd (ICARIA-MM): Haematological Abnormalities
ICARIA-MM: Haematological laboratory abnormalities in patients receiving SARCLISA® + Pd vs. Pd alone.2
The denominator used for the percentage calculation is the number of patients with at least 1 evaluation of the laboratory test during the considered observation period.1Adapted from SARCLISA® Summary of Product Characteristics.2
30.3% patients treated with SARCLISA® had neutropenic complications.2
- Patients with neutropenia should be monitored for signs of infection
- No dose reductions of SARCLISA® are recommended
- SARCLISA® dose delays and the use of colony-stimulating factors (e.g. G-CSF) should be considered to mitigate the risk of neutropenia
Full blood cell counts should be monitored periodically during treatment.2
Overall discontinuation rate did not increase with addition of SARCLISA® to Pd4
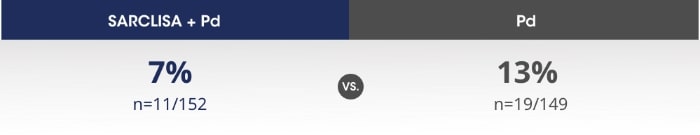
SARCLISA® + Pd (ICARIA-MM): Infusion-related reactions
ICARIA-MM: Infusion-related reactions* were mostly mild to moderate for patients receiving SARCLISA®.2
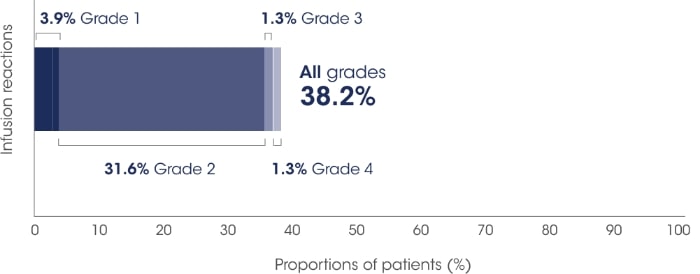
Adapted from SARCLISA®Summary of Product Characteristics.2
All patients who experienced infusion-related reactions, experienced them during the 1st infusion2

All patients who experienced infusion-related reactions, experienced them during the 1st infusion2

Infusion-related reactions were resolved on the same day in most patients, and all were reversible2

No delayed infusion-related reactions were observed and no mandatory post-infusion corticosteroid prophylaxis1,2
All patients who experienced infusion-related reactions, experienced them during the 1st infusion2
Infusion-related reactions were resolved on the same day in most patients, and all were reversible2
No delayed infusion-related reactions were observed and no mandatory post-infusion corticosteroid prophylaxis1,2
The most common symptoms of an infusion-related reaction included dyspnoea, cough, chills, and nausea. The most common severe signs and symptoms included hypertension and dyspnoea. The adverse event of severe anaphylactic reaction has been reported as a serious infusion reaction in 5 patients (0.3%) based on data from multiple clinical trials investigating isatuximab.2
In patients who experience grade 2 (moderate) infusion-related reactions, a temporary interruption in the infusion should be considered.2
*As reported by the investigator in the specific form of an adverse event, including infusion-related reaction, cytokine release syndrome, and drug hypersensitivity.1
Dosage
SARCLISA® + Pd (ICARIA-MM): Dosing and Administration
Recommended dosage2
10 mg/kg body weight administered as an IV infusion in combination with Pd*/250 mL fixed dosing volume.
Pre-medication† should be administered 15 to 60 minutes prior to infusion of SARCLISA®.
*For other products administered with SARCLISA®, refer to the respective current Summary of Product Characteristics.2
Infusion rates can increase over time, with a 75-minute infusion possible from the 3rd infusion onwards*2
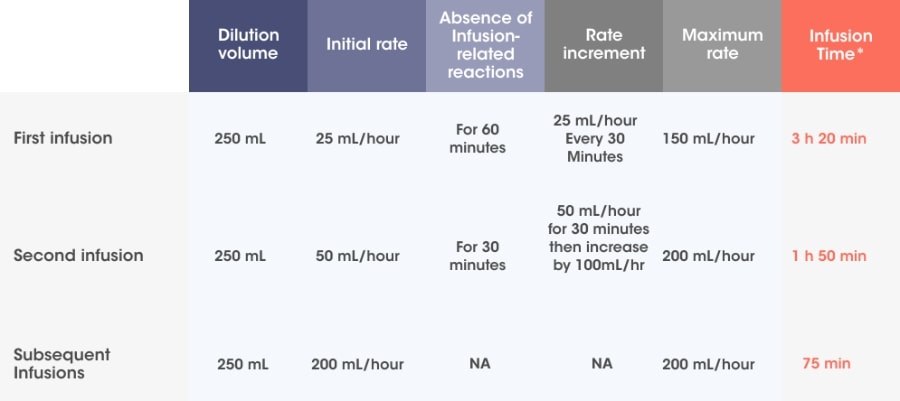
Adapted from SARCLISA® Summary of Product Characteristics.2
Incremental escalation of the infusion rate should be considered only in the absence of infusion-related reactions.2
- In patients necessitating an intervention (Grade 2, moderate infusion reactions), a temporary interruption in the infusion should be considered and additional symptomatic medicinal products can be administered. After symptom improvement to grade ≤1 (mild), SARCLISA® infusion may be resumed at half of the initial infusion rate under close monitoring and supportive care, as needed. If symptoms do not recur after 30 minutes, the infusion rate may be increased to the initial rate, and then increased incrementally.
- If symptoms do not resolve rapidly or do not improve to Grade ≤1 after interruption of SARCLISA® infusion, persist or worsen despite appropriate medicinal products, or require hospitalization or are life-threatening, treatment with SARCLISA ®should be permanently discontinued and additional supportive therapy should be administered, as needed.
*Infusion time possible in the absence of infusion-related reactions.2
† Pre-medication should be used prior to SARCLISA® infusion with the following medications to reduce the risk and severity of infusion-related reactions: dexamethasone 40 mg orally or IV (or 20 mg orally or IV for patients ≥75 years of age), paracetamol 650 mg to 1,000 mg orally (or equivalent), diphenhydramine 25 mg to 50 mg IV or oral (or equivalent [e.g. cetirizine, promethazine, dexchlorpheniramine]) (the IV route is preferred for at least the first 4 infusions).2
SARCLISA® + Pd (ICARIA-MM): Dosing Schedule
Dosing frequency decreases to Q2w in cycle 2.*2
Treatment is administered in 28-day cycles.2
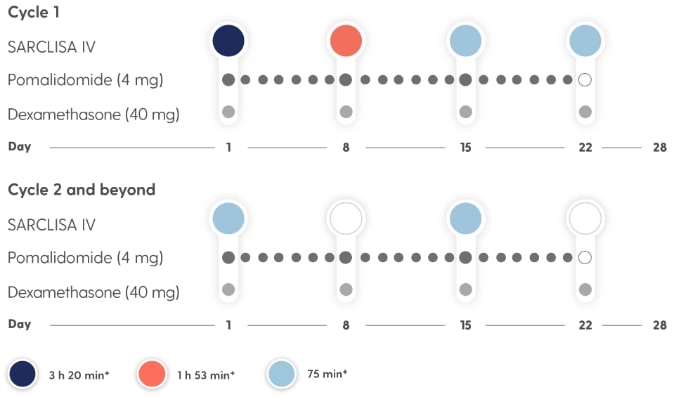
- Treatment is repeated until disease progression or unacceptable toxicity2
- For other medicinal products that are administered with SARCLISA®, refer to the respective current Summary of Product Characteristics2
- If a planned dose of SARCLISA® is missed, administer the dose as soon as possible and adjust the treatment schedule accordingly, maintaining the treatment interval2
*SARCLISA® dosing: 10 mg/kg body weight administered as an IV infusion in combination with Pd; fixed dosing volume of 250 mL.2
†Infusion time possible in the absence of infusion-related reactions.2
Infusion rates can increase over time, with a 75-minute infusion possible from the 3rd infusion onwards*2
Adapted from SARCLISA® Summary of Product Characteristics.2
Incremental escalation of the infusion rate should be considered only in the absence of infusion-related reactions.2
- In patients necessitating an intervention (Grade 2, moderate infusion reactions), a temporary interruption in the infusion should be considered and additional symptomatic medicinal products can be administered. After symptom improvement to grade ≤1 (mild), SARCLISA® infusion may be resumed at half of the initial infusion rate under close monitoring and supportive care, as needed. If symptoms do not recur after 30 minutes, the infusion rate may be increased to the initial rate, and then increased incrementally.
- If symptoms do not resolve rapidly or do not improve to Grade ≤1 after interruption of SARCLISA® infusion, persist or worsen despite appropriate medicinal products, or require hospitalization or are life-threatening, treatment with SARCLISA ®should be permanently discontinued and additional supportive therapy should be administered, as needed.
*Infusion time possible in the absence of infusion-related reactions.2
† Pre-medication should be used prior to SARCLISA® infusion with the following medications to reduce the risk and severity of infusion-related reactions: dexamethasone 40 mg orally or IV (or 20 mg orally or IV for patients ≥75 years of age), paracetamol 650 mg to 1,000 mg orally (or equivalent), diphenhydramine 25 mg to 50 mg IV or oral (or equivalent [e.g. cetirizine, promethazine, dexchlorpheniramine]) (the IV route is preferred for at least the first 4 infusions).2
SARCLISA® + Pd (ICARIA-MM): Dosing Schedule
Dosing frequency decreases to Q2w in cycle 2.*2
Treatment is administered in 28-day cycles.2
- Treatment is repeated until disease progression or unacceptable toxicity2
- For other medicinal products that are administered with SARCLISA®, refer to the respective current Summary of Product Characteristics2
- If a planned dose of SARCLISA® is missed, administer the dose as soon as possible and adjust the treatment schedule accordingly, maintaining the treatment interval2
*SARCLISA® dosing: 10 mg/kg body weight administered as an IV infusion in combination with Pd; fixed dosing volume of 250 mL.2
†Infusion time possible in the absence of infusion-related reactions.2
MOA
SARCLISA® is an anti-CD38 mAb with a specific target epitope5,6,7
CD38 is a transmembrane glycoprotein that has multiple functions, including enzymatic and receptor-mediated activity.5,6,7
MULTIMODAL ACTIONS:1,5,7–10
In vitro, SARCLISA®

Inhibits CD38 enzymatic activity through suppression of the CD38 ectoenzyme

Triggers myeloma cell death through tumour cell targeting, including ADCC, ADCP, and CDC

Enhances immune cell function through NK cell activation and a decrease in immuno-suppressors

Directly destroys myeloma cells through apoptosis, without the need for crosslinking
In vitro activity does not always correlate to clinical efficacy.
| SARCLISA® works synergistically with pomalidomide and dexamethasone in pre-clinical studies.5,7 |
AE, adverse event; ADA, anti-drug antibodies; AHG, antihuman globulin crossmatches; ASCT, autologous stem cell transplant; CDF, Cancer Drugs Fund; CI, confidence interval; COPD, chronic obstructive pulmonary disease; CR, complete response; DDT, dithiothreitol; ECOG, Eastern Cooperative Oncology Group; eGFR, estimated glomerular filtration rate; G-CSF, granulocyte-colony stimulating factor; HR, hazard ratio; Ig, immunoglobulin; IMWG, International Myeloma Working Group; IRC, independent response committee; ISS, International Staging System; IFE, immunofixation; IgG, immunoglobulin G; IQR, interquartile range; mAb, monoclonal antibody; MDRD, modification of diet in renal disease; MM, multiple myeloma; mPFS, median progression-free survival; M-Protein, monoclonal immunoglobulin; ORR, overall response rate; OS, overall survival; Pd, pomalidomide in combination with dexamethasone; PI, proteasome inhibitor; PFS, progression-free survival; PR, partial response; PK, pharmacokinetics; Q2w, once every two weeks. RBC, red blood cell; R-ISS, revised ISS; RRMM, relapsed and refractory multiple myeloma. sCR, stringent complete response; SPE, serum protein electrophoresis. VGPR, very good partial response. IV, intravenous.
References
- Attal M et al. Lancet 2019; 394: 2096–2107.
- SARCLISA®. Summary of Product Characteristics.
- NICE. Final appraisal document. Isatuximab with pomalidomide and dexamethasone for treating relapsed and refractory multiple myeloma October 2020. Available at: www.nice.org.uk/guidance/gid-ta10448/documents/final-appraisal-determination-document [accessed: April 2025].
- Attal M et al. Lancet 2019; 394: 2096–2107. Appendix.
- Richardson PG et al. Future Oncol 2018;14:1035–1047.
- Martin TG et al. Cells 2019; 8(12). pii:E1522.
- Jiang H et al. Leukemia 2016;30:399–408.
- van de Donk NWCJ et al. Blood 2018;131:13–29.
- Deckert J et al. Clin Cancer Res 2014;20:4574–4583.
- Moreno L et al. Clin Cancer Res 2019;25:3176–3187.
MAT-XU-2202578 (v7.0) Date of preparation: August 2025