- Wissen
- Quelle: Campus Sanofi
- 02.09.2024
Mukopolysaccharidose Typ 1 - Definition

Was ist MPS I
Mukopolysaccharidosen (MPS), wie z. B. Mukopolysaccharidose Typ 1 (MPS I), gehören zur Gruppe der lysosomalen Speicherkrankheiten (LSD, lysosomal storage disorders). LSD zählen zu den seltenen Erkrankungen. Insgesamt sind über 50 verschiedene LSD beschrieben. Dabei handelt es sich um Stoffwechselstörungen genetischen Ursprungs, die durch einen Mangel oder eine Defizienz von lysosomalen Enzymen verursacht werden. Hier erhalten Sie einen Überblick zu der Definition, der Vererbung und den Unterschieden zwischen den einzelnen Formen der Mukopolysaccharidosen.
Bei MPS dreht sich alles um GAGs (Glykosaminoglykane)
Glykosaminoglykane (GAG), früher auch als Mukopolysaccharide bezeichnet, sind komplexe Zuckermoleküle, deren Anreicherung der Erkrankungsgruppe der Mukopolysaccharidosen ihren Namen verleihen. Es gibt verschiedene Glykosaminoglykane, die zudem nicht in allen Organen und Geweben in gleichem Umfang vorkommen. Dies erklärt zum Teil auch die Unterschiede im klinischen Erscheinungsbild dieser Erkrankungen. Beispiele sind Dermatansulfat, Heparansulfat, Keratinsulfat, Chondroitinsulfat und Hyaluronsäure. Sie kommen physiologisch in zahlreichen Geweben wie der Haut, dem Bindegewebe, der Hornhaut sowie in weiteren Organen vor und übernehmen dort wichtige Funktionen. So dienen sie u. a. als strukturelle Komponente der extrazellulären Matrix oder sind als Signalmoleküle an der Modulation verschiedener zellulärer Prozesse beteiligt, wie z. B. Zellteilung, Zelldifferenzierung, Wachstum oder Zell-Zell-Kommunikation. So liegt es nahe, dass bei einer pathologischen Akkumulation dieser Mukopolysaccharide im Falle der MPS-Erkrankung die genannten Funktionen gestört sein können. Welches Glykosaminoglykan sich ansammelt, ist abhängig von dem spezifischen Enzym, das in seiner Funktion beeinträchtigt ist oder gänzlich fehlt (Abb. 1). Daraus ergeben sich dann die jeweils charakteristischen klinischen Merkmale, die aus der Akkumulation der jeweiligen Substrate entstehen (Clarke 2008, Sun 2018).
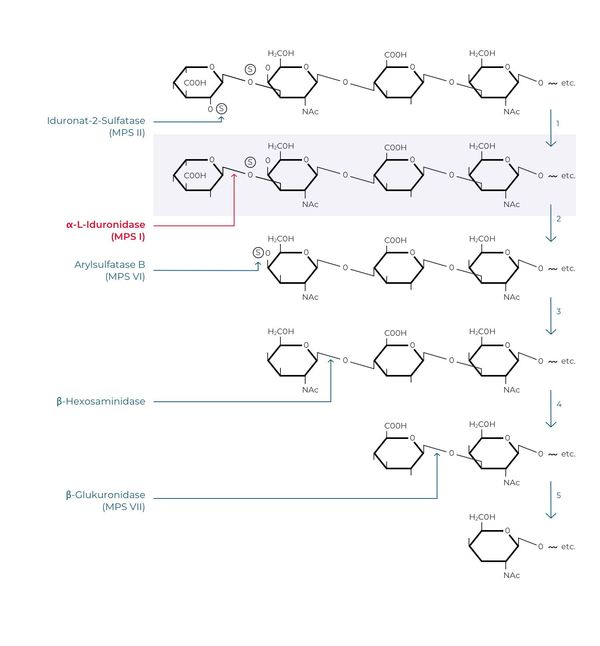
Abb.1: Enzymatischer Abbau von Glykosaminoglykanen am Beispiel von Dermatansulfat; modifiziert nach (Neufeld 2001).
Alpha-L-Iduronidase ist eine Exoglykosidase. Sie wird für den schrittweisen enzymatischen Abbau bestimmter Glykosaminoglykane (kurz: GAG), insbesondere von Dermatan- und Heparansulfat, benötigt. Glykosaminoglykane – früher auch als Mukopolysaccharide bezeichnet – sind Teil der extrazellulären Matrix und vor allem am Aufbau von Binde- und Stützgewebe beteiligt. GAG sind lange kettenförmige Moleküle, die in der Regel von zwei im Wechsel eingebauten, modifizierten Zuckerbausteinen gebildet werden. Der Mangel alpha-L–Iduronidase führt zur Behinderung des stufenweisen Katabolismus insbesondere der Glykosaminoglykane Dermatansufat und Heparansulfat, die sich in der Folge in den Lysosomen als Speichersubstanz anreichern. Direkt und indirekt kommt es in der Folge zu progredienten Fehlfunktionen auf Zell-, Gewebe- und Organebene (Clarke 2011). Durch das ubiquitäre Vorkommen von GAGs in der extrazellulären Matrix des Körpers sind nahezu alle Organsysteme – in Abhängigkeit der lokalen Bedeutung und Vorkommen von Dermatan- und Heparansulfat – in mehr oder minder großem Umfang beteiligt. Klinisch manifestiert sich dies insbesondere in Veränderungen des Hör- und Sehvermögens, des Skelett- und Stützapparates, Vergrößerungen der Organe (vor allem von Leber und Milz), Veränderungen der Atemwege und Lunge, der Herz-Kreislauffunktionen sowie bei schweren Verlaufsformen in einer Beteiligung des ZNS. Die Folge sind häufig frühe Behinderungen bis hin zum Tod der Patient*innen.
MPS – Lysosomen im Zentrum des Geschehens
Lysosomen spielen eine zentrale Rolle beim Abbau von zellulären Substraten. Der Abbau dieser Substrate wird von spezifischen lysosomalen Enzymen übernommen. Bei den Mukopolysaccharidosen sind bestimmte lysosomale Enzyme durch Mutationen nicht mehr oder nur noch eingeschränkt funktionsfähig. Infolge kommt es zu einer Akkumulation der entsprechenden Substrate in den Lysosomen, da diese durch die verminderte oder fehlende Enzymaktivität nicht mehr abgebaut werden können. Die Akkumulation der Substrate zeigt sich auf zellulärer Ebene durch eine Vergrößerung und Anreicherung von Lysosomen (Abb. 2) (Platt 2012, Sun 2018).
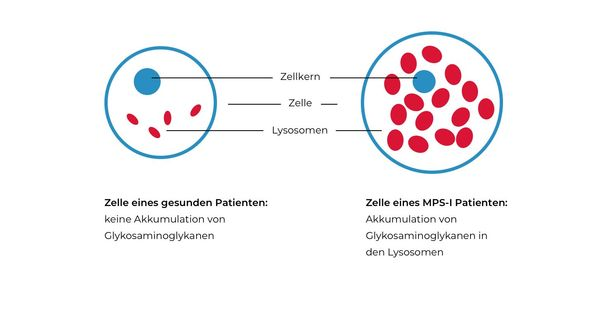
Abb.2: Pathologische Akkumulation von Substraten in den Lysosomen bei MPS.
Die Akkumulation der Substrate in den Lysosomen führt zunächst zu Zellschäden, die sich dann zu Gewebe- und in der Folge zu Organschäden ausweiten. Im Verlauf führen diese Veränderungen bei den Patienten zunehmend zu Symptomen (Abb. 3). Je nach Regenerationsfähigkeit des betroffenen Gewebes bzw. Organs sind diese Schäden zunehmend irreversibel. Um dies zu vermeiden, kommt einer möglichst frühen Diagnose und Therapie eine entscheidende Bedeutung zu (Clarke 2008, Sun 2018).
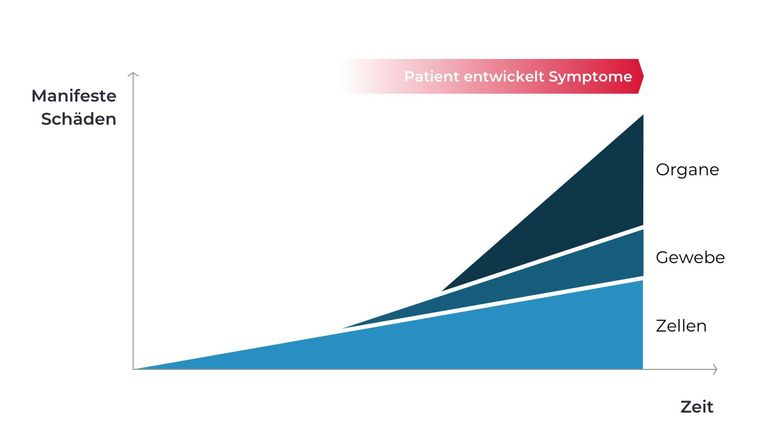
Abb.3: Schematische, vereinfachte Darstellung des Krankheitsverlaufs bei lysosomalen Speichererkrankungen.
Verlaufsformen der Mukopolysaccharidose Typ I (MPS I)
Es werden historisch 3 klinische Formen unterschieden:
- Morbus Hurler (MPS-I-H; ca. 60% der Patient*innen), die schwerste Form mit sehr früher Manifestation;
- Morbus Scheie (MPS-I-S; ca. 20%) mit verzögerter, milderer Ausprägung und
- Morbus Hurler-Scheie (MPS-I-H/S; ca. 20%) als intermediäre Verlaufsform
Da diese historische Einteilung der großen Breite an Variationen in der klinischen Symptomatik nicht gerecht wird, wird diese Einteilung heute zunehmend zugunsten einer Klassifikation in nicht-neuronopathische und neuronopathische MPS I verlassen.
| mit ZNS-Beteiligung | <----> | ohne ZNS-Beteiligung |
|---|---|---|
| schwere Verlaufsform Hurler MPS I-H | mittlere Verlaufsform Hurler-Scheie MPS I-H-S | verzögerte Verlaufsform Scheie MPS I-S |
| Deutliche geistige Retardierung | Normale oder fast normale Intelligenz | Normale Intelligenz |
| Verkürzte Lebenszeit | Normale Lebenszeit | |
| Skelettabnormitäten > | Hornhauttrübung | |
| Obstruktive Atemwegserkrankungen | Gelenksteifigkeit | |
| Progrediente und starke physische Probleme | Leichte bis mäßige physische Probleme | Weniger progressive physische Probleme |
Clarke LA. Expert Rev Mol Med 2008;10:e1
Clarke, LA, Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator, Rheumatology (2011) 50: Supplement 5 v13-v18
Neufeld E et al. (Hrsg.). McGraw-Hill, New York, 2001;3421-3452
Platt FM et al. The Journal of cell biology 2012;199(5):723-734
Sun A. Ann Transl Med 2018;6(24):476
Header-Foto: Sanofi-Aventis Deutschland GmbH MAT-DE-2303773-V2.0-12/2025
