- Wissen
- Quelle: Campus Sanofi
- 06.12.2024
Gaucher oder ASMD

Wichtige Tipps für die Praxis1

Thrombozytopenie und Splenomegalie können auch durch metabolische Ursachen wie die Stoffwechselkrankheiten Morbus Gaucher und ASMD bedingt sein.

Bei Morbus Gaucher und ASMD können folgende syndromale Befunde hinzukommen:
• Vergrößerter Oberbauch
• Erhöhte Blutungsneigung
• MGUS
• Häufig blaue Flecken
• Abgeschlagenheit
• Knochenschmerzen
• Atembeschwerden

Hämatoonkolog*innen spielen bei der Diagnose dieser Erkrankungen eine zentrale Rolle, da diese in der Allgemeinbevölkerung seltenen Krankheiten aufgrund ihrer Symptomatik im Patientengut der hämatologischen Praxis häufiger vorkommen.

Es sind spezifische Therapien verfügbar. Eine frühe Diagnose ist wichtig, um einen rechtzeitigen Therapiebeginn zu ermöglichen und irreversiblen Schäden vorzubeugen.

Bei Beachtung der differentialdiagnostischen Überlegungen kann mehr als jede 25. Testung zu einem positiven Nachweis einer lysosomalen Speichererkrankung führen.
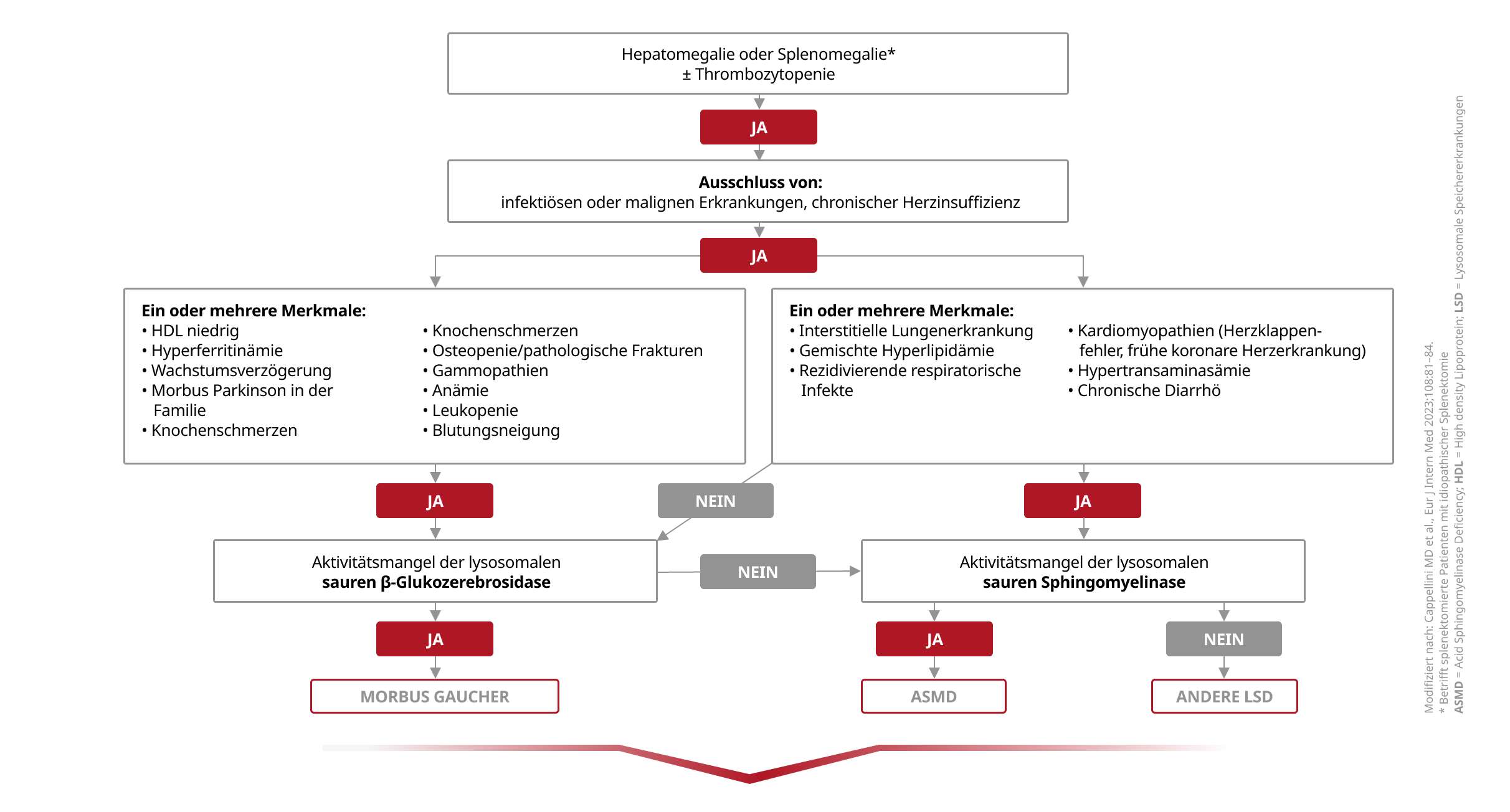
Dieser Diagnosealgorithmus sichert eine effiziente Abklärung der Verdachtsfälle:
Hinter einer Splenomegalie und/oder Thrombozytopenie können sich metabolische Erkrankungen wie Morbus Gaucher und ASMD verbergen. Da spezifische Therapien verfügbar sind, ist eine frühe Diagnose umso wichtiger.
Zur Diagnostik eignet sich unter anderem ein Trockenbluttest, der ASMD, Morbus Gaucher und LAL-D erfasst. Verschiedene Labore bieten solche Untersuchungen an. Neben lokalen Laboren an Universitätskliniken können das auch Anbieter von Großlaboren wie Amedes oder Archimed sein. Über das Kontaktformular dieser Seite erreichen Sie einen externen Service, der Informationen und mehr zum DBS Test von Archimed anbietet.
Was ist Morbus Gaucher?
Morbus Gaucher ist eine autosomal-rezessiv vererbte Stoffwechselerkrankung, bei der eine Splenomegalie und/oder Hepatomegalie, Veränderungen des Blutbildes sowie Knochenschmerzen auftreten können. Morbus Gaucher gilt als seltene Erkrankung und die Prävalenz wird mit 1–2 pro 100 000 angegeben (Ashkenazi: ca. 1 : 850). Die Ursache von Morbus Gaucher ist ein Mangel des lysosomalen Enzyms β-Glukozerebrosidase, wodurch sich das Stoffwechselprodukt Glukozerebrosid in Zellen des Monozyten-Makrophagen-Systems sammelt. Darum wird Morbus Gaucher zu den lysosomalen Speicherkrankheiten gezählt.1
Welche Symptomatik tritt bei Morbus Gaucher auf?
Morbus Gaucher ist eine multisystemische Erkrankung. Typisch sind Veränderungen im Blut, Organvergrößerungen und Schädigungen der Knochen. Deshalb werden meist folgende Untersuchungen vorgenommen:1
- Splenomegalie (85 % der Betroffenen) aufgrund der Infiltration der Milz mit Gaucher-Zellen und inflammatorischen Komponenten
-
Thrombozytopenie (68 %), meist moderat, aufgrund der Infiltration des Knochenmarks mit Gaucher-Zellen und Hypersplenismus
-
Hepatomegalie (63 %)
-
Anämie (34 %)
-
Knochenbeteiligung (radiologische Knochenbefunde 82 %, Knochenschmerzen 37 %, Knochenkrisen 7 %), trägt maßgeblich zur Morbidität bei
-
Abgeschlagenheit
-
Lungenbeteiligung (selten)
Wie wird Morbus Gaucher diagnostiziert?
Morbus Gaucher erfolgt durch den Nachweis einer verminderten Aktivität der β-Glukozerebrosidase und durch die Bestätigung von pathologischen Varianten im GBA-Gen. Überwiegend wird die Diagnose „Morbus Gaucher Typ 1“ im Kindesalter gestellt, bei einem Drittel der Betroffenen jedoch erst im Erwachsenenalter. Gründe hierfür können milde Verläufe, späte klinische Manifestationen sowie eine verzögerte Diagnose sein.1
Was ist ASMD?
Die Ursache von ASMD (Acid Sphingomyelinase Deficiency; früher Morbus Niemann-Pick Typ A, A/B und B) ist ein autosomal-rezessiv vererbter Mangel an dem lysosomalen Enzym saure Sphingomyelinase aufgrund von pathogenen Varianten im SMPD1-Gen. In der Folge kommt es zu einer Anreicherung von Sphingomyelin hauptsächlich in Zellen des Monozyten-Makrophagen-Systems, der Hepatozyten und in Kupffer-Zellen, bei schweren Verlaufsformen auch im zentralen Nervensystem. Sie schwellen zu sog. Schaumzellen an und sind häufig in Leber, Milz, Lunge, Lymphknoten, Nebennierenrinde und/oder im Knochenmark nachweisbar.1
Was sind die häufigsten Manifestationen bei ASMD?
Die häufigsten Manifestationen sind:1
- Splenomegalie (> 90 %)
-
Hepatomegalie (> 70 %)
-
interstitielle Lungenerkrankung (> 80 % radiologischer Befund)
-
Thrombozytopenie (> 50 %)
-
Abgeschlagenheit
-
außerdem können beteiligt sein: Nervensystem, Augen, Herz und Gefäße, Knochen und Gelenke, Magen und Verdauungssystem
Welche Verlaufsformen gibt es bei ASMD?
Die Geburtsprävalenz von ASMD beträgt 0,4–0,6/100 000. Die Verlaufsformen werden unterteilt in:1
- ASMD Typ A: infantil neuro-viszeral, schwerste Form, Beginn bald nach der Geburt, rasche Progredienz, Lebenserwartung: 2–3 Jahre
-
ASMD Typ B: chronisch-viszeral, Beginn überwiegend in der Kindheit, langsamere Progredienz, Lebenserwartung: Kindheit bis frühes Erwachsenenalter
-
ASMD Typ A/B: chronisch neuro-viszeral, intermediäre Verlaufsform mit variabler ZNS-Beteiligung
Wie wird ASMD diagnostiziert?
Die Diagnose von ASMD erfolgt durch den Nachweis der verminderten Aktivität der sauren Sphingomyelinase und durch die Bestätigung von pathologischen Varianten im SMPD1-Gen.

-
Bommer M. Differenzialdiagnose Thrombozytopenie und Splenomegalie; Praxis Report; Monat 2024, ISSN 1611-7891.
Header-Foto: Sanofi-Aventis Deutschland GmbH MAT-DE-2404527-2.0-10/2025
