- Wissen
- Quelle: Campus Sanofi
- 12.09.2024
Morbus Pompe Definition

Morbus Pompe ist eine seltene, autosomal-rezessiv vererbte Stoffwechselkrankheit aus der Gruppe der lysosomalen Speichererkrankungen. Sie äußert sich als progredient verlaufende Myopathie, die die Skelett-, Atem- und bei infantilem Beginn auch die Herzmuskulatur beeinträchtigt. Die Morbus Pompe Symptome und der Morbus Pompe Krankheitsverlauf sind individuell unterschiedlich und können zu andauernder Behinderung führen.1,2
Betroffene, die bereits im Säuglingsalter erkranken, versterben unbehandelt meist innerhalb des ersten Lebensjahres. Bei späterem Beginn der Erkrankung droht den Patient*innen die Abhängigkeit von Rollstuhl und Beatmungsgerät. Sie haben eine langfristig reduzierte Lebenserwartung.1-3Die Inzidenz des Morbus Pompe wird auf ca. 1:40.000-1:200.000 geschätzt.4
Ursache Enzymmangel
Die Erkrankung beruht auf einem genetisch bedingten Mangel oder dem völligen Fehlen des lysosomalen Enzyms saure α-1,4-Glukosidase (GAA, saure Maltase). In der Folge kommt es zu einer Akkumulation von Glykogen, vor allem in der Muskulatur; zusätzlich spielt eine gestörte Autophagie bei der Pathogenese eine Rolle.1,2,4
In der Folge werden zunächst einzelne Zellen und zunehmend im Verlauf das gesamte Muskelgewebe in seiner Funktion beeinträchtigt.1,2,4 Einmal eingetretene Schäden sind meist irreversibel.5
Morbus Pompe ist somit eine Glykogenspeichererkrankung und zählt zu den hereditären metabolischen Myopathien.1Er ist unter anderem auch als saurer Maltase-Mangel (englisch: Acid Maltase Deficiency, AMD), α-1,4-Glukosidase-Mangel, saurer α-Glukosidase-Mangel oder Glykogenose Typ II bekannt. Der ICD10 Code für die Diagnose Morbus Pompe lautet E74.02.
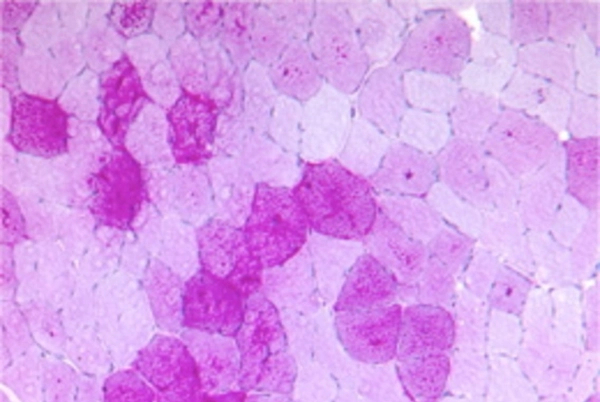
Abb.: Nachweis vermehrter Glykogeneinlagerung in einigen Muskelfasern (PAS-Färbung des M. vastus lateralis links). (Bildquelle: Institut für Neuropathologie, Universitätsklinik Mainz)

Abb.: Krankheitsprogression bei lysosomalen Speicherkrankheiten wie Morbus Pompe im Modell
Therapie
Seit die erste Enzymersatztherapie im Jahr 2006 zugelassen wurde, sind weitere spezifische Therapien, z.B weiterentwickelte Enzymersatztherapien oder Gentherapien, zugelassen worden oder aktuell noch in der klinischen Entwicklung. Zusätzlich bleibt eine symptomatische Morbus Pompe Begleittherapie unverzichtbar.
Erfahren Sie mehr über:
Häufig gestellte Fragen zur Definition von Morbus Pompe
Was ist Morbus Pompe?
Morbus Pompe ist eine seltene, autosomal-rezessiv vererbte Stoffwechselkrankheit aus der Gruppe der lysosomalen Speichererkrankungen. Sie äußert sich als progredient verlaufende Myopathie, die die Skelett-, Atem- und bei infantilem Beginn auch die Herzmuskulatur beeinträchtigt. Unbehandelt kann sie zu dauernder Behinderung oder zum Tod führen. Die weltweite Inzidenz wird auf ca. 1:40.000–1:300.000 geschätzt1,2,4
Was ist die Ursache von Morbus Pompe auf molekularer Ebene?
Morbus Pompe wird durch einen genetisch bedingten Mangel oder das völlige Fehlen des lysosomalen Enzyms saure α-1,4-Glukosidase (GAA, saure Maltase) verursacht. Infolgedessen kommt es zur Akkumulation von Glykogen, vor allem in Muskelzellen, was eine gestörte Autophagie auslöst. Einmal eingetretene Schäden sind meist irreversibel.1,2,5
Welche Therapieoptionen gibt es bei Morbus Pompe?
Seit der ersten Enzymersatztherapie (ERT) im Jahr 2006 stehen spezifische Therapien zur Verfügung, darunter weiterentwickelte Enzymersatztherapien und Gentherapien. Zusätzlich ist eine symptomatische Morbus-Pompe-Begleittherapie (Atemtherapie, Physiotherapie, Ernährung) unverzichtbar.1,2
-
Hirschhorn R, Reuser AJJ. McGraw Hill, 2001:3389-3420
-
Kishnani PS et al. Genet Med 2006; 8: 267-288
-
Hagemans ML et al. Neurology 2004; 63: 1688-1692
-
Kohler N et al. Neurotherapeutics 2018; 15: 928-942
-
Hundsberger T et al. Schweiz Arch Neurol Psychiat 2010; 161:55–59
Header-Foto: Sanofi-Aventis Deutschland GmbH MAT-DE-2302548 - v3.0 - 06/2026
