
Fabry Request Information
Learn more
This website contains promotional content and is intended for Healthcare Professionals based in the United States only.
Understand Fabry disease inheritance1,2
Fabry disease is a genetic disorder that can present in families for years before being accurately diagnosed. For every index patient diagnosed, an average of 5 additional affected family members may be identified. Fabry disease is progressive, so early diagnosis is important.
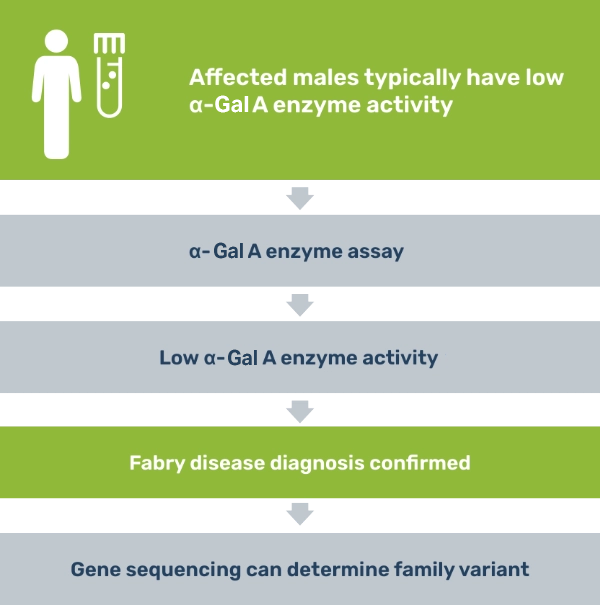
Testing is straightforward3
A definitive diagnosis can be confirmed for males through an α-Gal A enzyme assay and for females through GLA gene sequencing.
Multi-systemic symptoms require multi-disciplinary care4
Patients may present with a range of symptoms that may require management and monitoring from a variety of specialists.
Fabry disease is an X-linked lysosomal storage disorder that affects men, women, and children of all ethnicities.3,5 The prevalence of Fabry disease is estimated to be 1:40,000 in males and 1:20,000 in females.2,3
Fabry is a progressive, multisystemic disorder that can result in potentially life-threatening damage to the kidney, heart, and brain.1,5
In Fabry disease, variants in the GLA gene, located on the X chromosome, result in defects in the synthesis and/or function of α-galactosidase A (α-Gal A).6
α-Gal A is a lysosomal enzyme, which usually metabolizes globotriaosylceramide (also known as GL-3 or Gb3) and prevents it from accumulating.7
Complete or partial deficiency of α-Gal A leads to progressive accumulation of glycosphingolipids, particularly GL-3, in the lysosomes of numerous cell types.8
In Fabry disease, accumulation of GL-3 begins prenatally, or in utero, and continues over a lifetime.3,5 With advancing age, the progressive lysosomal GL-3 accumulation—particularly in the microvasculature—leads to renal failure, heart disease, stroke, and premature demise, typically in the fourth or fifth decade of life.1,7,8,9
Behind every patient diagnosed with Fabry is a family that is at risk. For every index patient diagnosed, an average of 5 additional affected family members may be identified.2
Over 900 variants in the GLA gene associated with Fabry disease have been identified.6,10 Notably, most families with classic disease have variants specific to their family.5

As an X-linked disease, the genetic variant that causes Fabry disease can be passed on by both males and females, and females are not simply carriers.3,8,11 Men have a 100% chance of passing the altered gene to their daughters and a 0% chance of passing it to their sons, while women with Fabry disease have a 50% chance of passing the altered gene to each daughter and son.8
Females with the altered GLA gene can develop Fabry, but the manifestations can be heterogeneous because of organ-and-tissue-specific X-inactivation.
For every index patient diagnosed, an average of 5 additional affected family members may be identified.2
Fabry disease is a genetic disorder; consequently, genetic counseling and family screening are important components of familial risk assessment.8
A positive family history is a strong indicator for Fabry disease, however de novo or spontaneous variants have been documented.3,12 Consequently, absence of a family history does not rule out a diagnosis of Fabry disease.3,12
The age of presentation of Fabry disease is variable, as are the presenting symptoms and the clinical course. A subset of disease symptoms usually appear during childhood.13 However, the disease often goes unrecognized until adulthood, when more severe symptoms prompt patients to pursue diagnosis.
Early signs and symptoms of Fabry disease, which can start as early as childhood or adolescence, may include pain, gastrointestinal disturbances, angiokeratomas, hypohidrosis, proteinuria, and other signs and symptoms.8 As Fabry disease progresses, renal function deterioration and hypertrophic cardiomyopathy may occur, and the risk of developing fatal complications such as end-stage renal disease, stroke, cardiac fibrosis, arrhythmias, and premature death increases.3,8

A definitive diagnosis can be made in males by testing for deficient α-galactosidase A (α-Gal A) enzyme activity in plasma, leukocytes, cultured skin fibroblasts, biopsied tissue, or dried blood.3
Because females with Fabry disease can have α-Gal A activity in the low to normal range in plasma, the patient should receive DNA analysis (either variant analysis or linkage analysis, depending on whether the family variant is known).3 Though an enzyme assay is not required for diagnosis, results can be helpful for understanding genotype-phenotype correlations.
If you suspect Fabry disease in a patient, testing is a straightforward process.3
.png)
Although females with a variant in the GLA gene may be asymptomatic or present with mild clinical manifestations, definitive identification of heterozygotes is important. Diagnosis allows practitioners to monitor for new or worsening symptoms and can help identify other family members with the disease.3
Monitoring patients with Fabry disease by routinely assessing their condition can help monitor development of new symptoms and disease progression, prompting timely disease management.9 The Fabry Registry Board of Advisors has developed a minimum schedule of assessments for patients both under 18 years of age and over 18 years of age. The Fabry Registry is sponsored and administered by Sanofi.
Routine monitoring for new or worsening symptoms is key to prompt disease management9
Routine monitoring of females with Fabry is particularly important. Patients may be asymptomatic initially, but may eventually develop symptoms over time.17
| Upon Enrollment | Every 6-12 monthsa | Every 24-36 months | At the time of an event or therapy change | |
| GENERAL | ||||
| Medical History, with particular focus on: | ■ | ■ | ■ | |
| ||||
| ||||
| ||||
| ||||
| Family History | ■ | ■ | ||
| Physical Exam | ■ | ■ | ■ | |
| Vital Signs, Height and Weight | ■ | ■ | ■ | |
| Blood Pressureb | ■ | ■ | ■ | |
| Enzyme Activity and Genotype | ■ | |||
| Disease-Specific Therapy Status | ■ | ■ | ■ | |
| Concomitant Medication Assessment | ■ | ■ | ■ | |
| Pediatric Quality of Life Assessment - PedsQLTM Pediatric Quality of Life Inventory | ■ | ■ | ■ | |
| Pediatric Quality of Life Assessment - PedsQLTM Multidimensional Fatigue Scale | ■ | ■ | ■ | |
| Pediatric Pain Assessment - PedsQLTM Pediatric Pain QuestionnaireTM | ■ | ■ | ■ | |
| LABORATORY TESTS | ||||
| Glomerular Filtration Rate (GFR)c | ■ | ■ | ■ | |
| Albuminuria and Proteinuriad | ■ | ■ | ■ | |
| OTHER STUDIES | ||||
| Audiologic Evaluatione | ■ | ■ | ■ | |
| Cranial MRI - T1, T2 and FLAIR | ■ | ■f | ■f1 | |
| Electrocardiogramg | ■ | ■ | ■ | |
| Echocardiogramh | ■ | ■ | ■ | |
| Cardiac MRIi | ■ | ■ | ■ | |
| Opthalmology - Slit Lamp Examj | ■ | ■ |
The Recommended Schedule of Assessments represents the core Fabry disease-related assessments that allow evaluation of a patient's disease progression over time. Physicians will determine the actual frequency of necessary assessments according to a patient's individualized need for medical care and routine follow-up. This is not a comprehensive list of recommended assessments. For additional information, please contact your Sanofi Representative.
These recommendations were developed by the Fabry Registry Board of Advisors, a group of physicians who have experience in managing patients with Fabry disease. The Fabry Registry is sponsored and administered by Sanofi
* Initiation of Laboratory Tests, Imaging, and Other Studies: There is variability in the clinical complications and progression of Fabry disease. Children are at risk for life-threatening complications. There are no biomarkers available to discern mildly affected from severely affected patients. In children with a family history of early presenting or severe disease, complete evaluations should be done at the time of diagnosis. Other patients should be completely evaluated at no later than 5 years of age.
a Patients are recommended to undergo these evaluations every 6 months; for those with milder disease, once per year may be sufficient.
b Blood pressure should be measured 3 times at each assessment; only the last 2 measurements should be recorded.
c GFR should be measured directly every 24-36 months until age 15, and annually thereafter. If direct measurement is not possible, serum creatinine levels should be obtained at the recommended intervals for an estimation of GFR, which is a less sensitive method.
d First morning voided urine for protein, albumin, and creatinine in order to calculate a protein/creatinine ratio and albumin/creatinine ratio. Protein, albumin, and creatinine measurements can also be performed on timed samples (e.g., 24 hours).
e Audiologic evaluation should be performed at the earliest age that is practical.
f Cranial MRIs should be performed at ages 10, 15, and 18 years.
f1 At the time of a cerebrovascular event, a cranial MRI should also include diffused weighted images and apparent diffusion coefficient (DWI/ADC).
g Electrocardiogram should be performed starting at age 10-12 years. If abnormal and/or clinical symptoms arise, Holter monitoring is recommended.
h Echocardiogram should be performed starting at age 10-12 years.
i Cardiac MRI is recommended to be performed in patients under age 25 if cardiac hypertrophy significant arrhythmia is present.
j Monitor yearly if retinal vessel tortuosity is noted.
| Upon Enrollment | Every 6 months | Every 12 months | Every 24-36 months | At the time of an event or therapy change | |
| GENERAL | |||||
| Medical History | ■ | ■ | ■ | ||
| Family History | ■ | ■ | |||
| Physical Exam | ■ | ■ | ■ | ||
| Vital Signs, Height and Weight | ■ | ■ | ■ | ||
| Enzyme Activity and Genotype | ■ | ||||
| Disease-Specific Therapy Status | ■ | ■ | ■ | ||
| Concomitant Medication Assessment | ■ | ■ | ■ | ||
| Quality of Life (SF-36°, BPI) | ■ | ■ | ■ | ||
| LABORATORY TESTS | |||||
| Serum Creatininea and BUN | ■ | ■ | ■ | ||
| Urine Protein Excretionb | ■ | ■ | ■ | ||
| Lipid panel | ■ | ■ | |||
| OTHER STUDIES | |||||
| Audiologic Evaluation | ■ | ■ | ■ | ||
| Cranial MRI - T1, T2 and FLAIR | ■ | ■ | ■c | ||
| Electrocardiogramd | ■ | ■ | ■ | ||
| Echocardiogram | ■ | ■ | ■ | ||
| 24-Hour Holter Monitoring | ■ | ■ | ■ | ||
| Cardiac MRIf | ■ | ■f1 | ■f1 | ■f2 | |
| Respiratory - Spirometry Examg | ■ | ■ | |||
| Ophthalmology - Slit Lamp Examh | ■ |
The Recommended Schedule of Assessments represents the core Fabry disease-related assessments that allow evaluation of a patient's disease progression over time. Physicians will determine the actual frequency of necessary assessments according to a patient's individualized need for medical care and routine follow-up.
a Directly measuring glomerular filtration rate (GFR) is recommended if a more precise evaluation is desired.
b 24-hour or first morning void urine for protein, creatinine, and albumin.
c At the time of an event, a cranial MRI should also include DWI/ADC.
d If electrocardiogram is abnormal and/or clinical symptoms arise, Holter monitoring is recommended.
e Annual 24-hour Holter monitoring is recommended for males 30 years of age or older and females 40 years of age or older.
f Cardiac MRI is recommended at Fabry diagnosis for patients ages 25 and older. It is recommended to be performed under age 25 if cardiac hypertrophy or significant arrhythmia is present.
f1 If first MRI is abnormal: 1) patients with moderate or severe LVH receiving ERT should have MRI annually; 2) patients with significant arrhythmia should have MRI at least every 2 years or at frequency factoring cardiac disease severity and the physician's clinical judgment; 3) males with no or mild LVH zreceiving ERT should have MRI every 2 years.
f2 If first MRI is normal, repeat every 5 years or earlier if ECG/ECHO results are abnormal on annual exam.
g If spirometry is abnormal, perform yearly.
h Monitor yearly if retinal vessel tortuosity noted.
Have a question or want additional information? A Sanofi representative is available to answer your disease- or product-related questions. Click the link below to complete a request.
|
Symptoms and diagnosis |
Inheritance |
For Cardiologists |
For Nephrologists |
Monitoring |
Sanofi’s Medical Information Department can provide information on diagnostic testing, pharmacovigilance/safety, and Fabry disease.
|
Visit our Medical Information Website Please call 8 AM until 6 PM EST, Monday through Friday: 1-800-745-4447, option 2 (toll-free)/ |
CareConnect is a free, voluntary and confidential support program for eligible patients and families living with certain lysosomal storage disorders (LSDs)
Connected Education: Comprehensive disease education from diagnosis and beyond for individuals, families, and communities.
Connected Team: Experts who connect the dots between specialists, insurance, and appointments for a less fragmented care experience.
Connected Experience: Programs designed to support patients by connecting them with experts and the community to navigate life transitions and manage treatment.
If affording treatment is an issue, CareConnect may be able to help eligible patients access financial assistance. To learn more about our range of support offerings, connect with us careconnectpss.com/hcp, call 1-800-745-4447 option 3, or email info@careconnectpss.com
.svg)
Fabry Support and Information Group
www.fabry.org
660-663-1355
National Fabry Disease Foundation (NFDF)
www.fabrydisease.org
800-651-9131
.svg)
National Kidney Foundation
www.kidney.org
855-653-2273
National Organization for Rare Disorders (NORD)
www.rarediseases.org
800-999-6673
.svg)
American Kidney Fund
www.kidneyfund.org/fabry
800-638-8299
Sanofi is committed to providing you with materials to help educate your patients about lysosomal storage disorders. A variety of patient educational materials are available to help patients understand Fabry disease, its symptoms, and management.
The Fabry Registry is an ongoing, observational database that tracks the natural history and outcomes of patients with Fabry disease. The Fabry Registry contains data from over 5000 patients from more than 50 countries worldwide. All Fabry disease patients are eligible for enrollment irrespective of their treatment status, and all physicians managing patients with Fabry disease are encouraged to participate in the Fabry Registry.
The Fabry Registry is a global outcomes assessment and disease management program that compiles patient outcomes data from routine clinical practice to provide the medical community with resources to help optimize patient care. The Fabry Registry provides a repository that allows for the exchange of information and aggregate data to facilitate clinical decision-making and data reports, and serves as a vehicle for collaborative studies.
For more information on the Fabry Registry, please visit the Registry website or call toll-free
800-745-4447, ext. 15500
.svg)

References: 1. Eng CM et al. J Inherit Metab Dis. 2007;30(2):184-192. 2. Laney DA et al. J Genet Couns. 2008;17(1):79-83. 3. Desnick RJ et al. In: The Online Metabolic and Molecular Bases of Inherited Diseases. Accessed June 21, 2025. doi:10.1036/ommbid.181 4. Peters FPJ et al. Postgrad Med J. 1997;73(865):710-712. 5. Desnick RJ et al. Ann Intern Med. 2003;138(4):338-346. 6. Nagueh SF. Circulation. 2014;130(13):1081-1090. 7. Ortiz A et al. Nephrol Dial Transplant. 2008;23(5):1600-1607. 8. Germain DP. Orphanet J Rare Dis. 2010;5(30):1-49. 9. Sims K et al. Stroke. 2009;40(3):788-794. 10. The Human Gene Mutation Database. Institute of Medical Genetics in Cardiff. Accessed July 8, 2025. Available at: http://www.hgmd.cf.ac.uk/ac/index.php. 11. Wang RY et al. Genet Med. 2007;9(1):34-45. 12. Redonnet-Vernhet I et al. J Med Genet. 1996;33(8):682-688. 13. Shelley ED et al. Pediatr Dermatol. 1995;12(3):215-219. 14. Barba-Romero MA et al. Int J Clin Pract. 2011;65(8):903-910. 15. Mehta A. QJM. 2002;95(10):647-653. 16. Wilcox WR et al. Mol Genet Metab. 2008;93(2):112-128. 17. Hopkin RJ et al. Mol Genet Metab. 2016;117(2):104-113. 18. Fabry Registry Recommended Schedule of Assessments 2015. Accessed July 8, 2025. Available at: https://www.fabrydisease.org/images/ReferencePDFs/fabry-registry-schedule-of-assessments.pdf.
MAT-US-2210037-v3.0-8/2025
