- Artikel
- Bron: Campus Sanofi
- 9 feb 2023
Algemene informatie ziekte van Pompe

Wat is de ziekte van Pompe?
De ziekte van Pompe is een autosomaal recessief erfelijke spierziekte, behorend tot de groep lysosomale stapelingsziekten. De ziekte van Pompe was de eerste lysosomale stapelingsziekte die als zodanig werd beschreven (in 1932, door de Nederlandse patholoog J.C. Pompe).
Er worden drie vormen van deze ziekte onderscheiden: een klassieke, vroeg-infantiele variant, een niet-klassieke infantiele variant en een variant met een latere ziekte-aanvang.
De klassieke, vroeg-infantiele variant is de ernstigste vorm, met een snel progressief verloop. Bij deze vorm staan hypotonie en cardiomyopathie op de voorgrond. De andere twee varianten hebben een milder verloop, zijn minder progressief en hebben als belangrijkste kenmerk progressieve spierzwakte.
Hoe ontstaat de ziekte van Pompe?
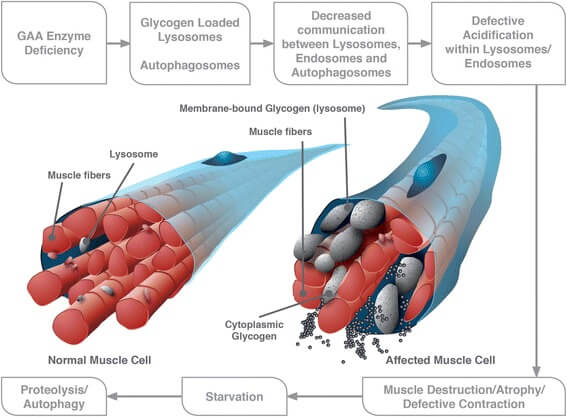
Deze aandoening wordt veroorzaakt door mutaties in het GAA-gen; hierdoor ontstaat een deficiëntie van zure-α-glucosidase, ook wel zure maltase genoemd, een enzym dat lysosomaal glycogeen afbreekt tot glucose. Als gevolg van deze enzymdeficiëntie treedt accumulatie van glycogeen op in vooral hart- en skeletspiercellen en gladde spiercellen, maar ook in cellen van andere lichaamsweefsels, waaronder die van de lever en van het zenuwstelsel. De stapeling van glycogeen leidt tot opzwelling en ruptuur van de lysosomen met verlies van glycogeen in het cytoplasma; hierdoor treedt celschade op.
Fig.1 Zowel opgezwollen en gescheurde lysosomen als overmatige autofagie dragen bij tot de progressieve myopathie in de varianten van de ziekte van Pompe met latere aanvang. [Bron: BMC Neurology 2015; 15: 205 Uit: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4608291/bin/12883_2015_412_Fig1_HTML.jpg
Symptomen
Het klinische spectrum van de ziekte van Pompe strekt zich uit van een snel progressieve, snel fatale vroeg-infantiele vorm met hypertrofische cardiomyopathie en myopathie van de skeletspieren tot mildere vormen met een latere ziekte-aanvang (tot op volwassen leeftijd), die gekenmerkt worden door progressieve myopathie, veelal zonder significante cardiomyopathie.
De ernst van de klinische presentatie, het type weefsel wat aangedaan is en de leeftijd van ziekte-aanvang correleren over het algemeen met de specifieke genmutatie en de mate van overgebleven enzymactiviteit.
De niet-klassieke infantiele vorm en de vormen met een latere ziekte-aanvang resulteren in immobiliteit en, indien onbehandeld, voortijdig overlijden als gevolg van respiratoir falen.
Diagnose
De infantiele variant
Als er bij een baby sprake is van hypertrofische cardiomyopathie, musculaire hypotonie en matig verhoogde CK-waarden in het bloed, is dat een sterke aanwijzing dat het kind de ziekte van Pompe heeft. Een vroege diagnose is van groot belang.
De varianten met een latere ziekte-aanvang
De diagnostiek van de varianten met een latere ziekte-aanvang kan veel tijd in beslag nemen, omdat deze qua klinische presentatie een grote gelijkenis vertonen met limb-girdle spierdystrofie (LGMD). Als in een vroeg stadium ook sprake is van een verzwakt diafragma en respiratoire klachten als gevolg daarvan, en eventueel ook hartritmestoornissen of vasculaire problematiek, kan dat een extra aanwijzing zijn in de richting van de ziekte van Pompe.
Omdat er bij de varianten van de ziekte van Pompe met een latere ziekte-aanvang geen sprake is van een specifiek klinisch fenotype, is het belangrijk de ziekte van Pompe te includeren in de differentiaaldiagnostiek van LGMD.
DNA analyse
Informatie over de mutatie in het GAA-gen die bij een individu de ziekte van Pompe veroorzaakt, is onder andere van belang voor prognostisch familieonderzoek. Mede om die reden zal, als de ziekte van Pompe is vastgesteld aan de hand van enzymdiagnostiek, in de meeste gevallen ook een DNA-analyse verricht worden.
Enzymdiagnostiek
De diagnose kan worden bevestigd door middel van bepaling van GAA-activiteit in bloed (leukocyten).
Prenatale diagnostiek
Bij het ongeboren kind kan zowel de enzymatische diagnose als de DNA-diagnose worden gesteld door middel van chorionvillusbiopsie (vlokkentest) en amniocentese (vruchtwaterpunctie). Voor de klassieke infantiele vorm is prenatale enzym diagnostiek een betrouwbare methode om de ziekte van Pompe aan te tonen of uit te sluiten. Voor de mildere varianten van de ziekte van Pompe geldt dat niet altijd: in die gevallen kan DNA-analyse nodig zijn om definitief uitsluitsel te geven. Voor het kunnen verrichten van prenatale DNA-diagnostiek moeten in principe de in de desbetreffende familie voorkomende DNA-mutaties in het GAA-gen bekend zijn.
Soorten behandelingen
Tot nog toe is de ziekte van Pompe niet te genezen. Wel is er een behandeling mogelijk waarbij het enzym tekort wordt gecompenseerd met van buitenaf toegediend vervangend enzym (ERT). Daarnaast kan de ziekte vooralsnog uitsluitend (en waar mogelijk) symptomatisch behandeld worden.
Sinds 2006 is voor de behandeling van de ziekte van Pompe enzymvervangende therapie beschikbaar, welke door middel van een infuus in de bloedbaan van patiënten gebracht wordt. Tot op heden is ERT de enige behandelmethode voor de ziekte van Pompe.
Aanvullende informatie

De plek om alle lysosomale stapelingsziekten te vinden
Complete informatie per stapelingsziekte op 1 plek. Of u nu zoekt op symptoom
of op naam, op lyso.nl wordt u verder geholpen.

Antwoord op vragen over de ziekte van Pompe
Uitgebreide informatie om te onder-steunen met de uitdagingen die de ziekte van Pompe met zich mee kan brengen.
De plek om alle lysosomale stapelingsziekten te vinden
Complete informatie per stapelingsziekte op 1 plek. Of u nu zoekt op symptoom
of op naam, op lyso.nl wordt u verder geholpen.
Antwoord op vragen over de ziekte van Pompe
Uitgebreide informatie om te onder-steunen met de uitdagingen die de ziekte van Pompe met zich mee kan brengen.
Wat is een lysosomale stapelingsziekte?
Bekijk hier een algemene uitleg wanneer er sprake is van lysosomale stapelingsziekte en wat er gebeurt in de cellen.
Contact
