%20(1)%20(1).2026-02-05-13-37-57.svg)
How ASMD Can Impact Quality of Life and Average Life Expectancy
Learn more
This website contains promotional content and is intended for Healthcare Professionals based in the United States only.
A progressive genetic disease with multisystemic complications and varying severity
ASMD (acid sphingomyelinase deficiency) can result in severe multisystemic complications, including morbidity and early mortality.1,2
Early diagnosis of ASMD is imperative
Symptoms of ASMD are variable and often overlap with other conditions, leading to frequent diagnostic delays and missed diagnoses.1-3
Include ASMD and Gaucher disease in your differential diagnosis and parallel test
With patients experiencing average diagnostic delays of ~5 years, parallel testing can help facilitate timely diagnosis and symptom management.2,3,a
aBased on a prospective, cross-sectional survey of 59 ASMD type B patients.
ASMD (acid sphingomyelinase deficiency), historically known as Niemann-Pick disease Types A, A/B, and B, is a genetic disease with progressive and multisystemic symptoms that can lead to early mortality. The disease is caused by deficiency of the enzyme acid sphingomyelinase (ASM), resulting in buildup of the substrate sphingomyelin in cells. Accumulation of sphingomyelin impacts major organs, which may include the liver, lungs, spleen, blood and gastrointestinal system.1
aBased on a prospective, cross-sectional survey of 59 ASMD type B patients
| TYPE A | TYPE A/B | TYPE B (most commonb) | |
| Onset | Early infancy | Infancy to childhood | Infancy to adulthood |
| Phenotype | Rapidly progressive, severe multiorgan involvement and neurodegeneration | Variably progressive, variable multiorgan disease and neurodegeneration | Slower progressive, variable multiorgan disease with little to no neurological involvement |
| Life expectancy | 2 to 3 years of age | Childhood to mid-adulthood | Childhood to late adulthood |
bBased on patient population from a multicenter, historical cohort study (N=100).
Regardless of ASMD type, deficient ASM activity can lead to lifelong, multisystemic complications.2
ASMD is caused by pathogenic variants in the SMPD1 gene – which encodes acid sphingomyelinase (ASM) – resulting in ASM enzyme deficiency.2
.jpg)
ASM breaks down sphingomyelin in the lysosomes of the cell.2
Sphingomyelin plays an important role in cellular processes, such as cell cycle regulation, cell signaling, and apoptosis.5,6
Decreased ASM activity leads to buildup of sphingomyelin.1
Lysosomal accumulation of sphingomyelin in the monocyte-macrophage system causes damage that can result in life-threatening, multiorgan complications.1



aSymptom prevalence data for splenomegaly, interstitial lung disease, hepatomegaly, thrombocytopenia, and pediatric growth delay are only for patients with ASMD type B. Gastrointestinal issues symptom prevalence is for all ASMD types.1,4
Symptoms of ASMD often overlap those of other lysosomal storage diseases, hematologic malignancies, and diseases of the heart and lungs. As a result, missed diagnoses and diagnostic delays are common.2
Conditions that share the hallmark signs and symptoms of ASMD type B include Gaucher disease type 1, acute lymphoblastic leukemia, non-Hodgkin lymphoma, chronic hepatitis B, cystic fibrosis, and congestive heart failure.2
Gaucher disease – another rare lysosomal storage disease – shares significant phenotypic overlap with ASMD. Similar to ASMD, Gaucher disease is characterized by multisystemic and progressive symptoms that vary in onset and clinical presentation.2,9

The dots in the table above represent the most commonly presenting symptoms in each of the respective diseases (ASMD type B and Gaucher disease type 1).2,3,9-11,14
bThis symptom is not seen as commonly in Gaucher disease type 1.
ASMD symptoms vary in onset and clinical presentation, and often overlap with those of more commonly seen and considered conditions. Due to this variability and symptom overlap, diagnostic delays are common.1,2,10
Diseases of the heart, liver, lungs, and hematologic malignancies may exhibit symptoms similar to ASMD. Another lysosomal storage disease, Gaucher disease, shares significant symptom overlap with ASMD.2
aBased on a prospective, cross-sectional survey of 59 ASMD type B patients.
Splenomegaly and hepatomegaly are often the first presenting signs of ASMD and Gaucher disease. Further evaluations may reveal other compounding symptoms that should prompt diagnostic testing.2

HDL-C=high-density lipoprotein cholesterol.
Adapted from McGovern MM et al. Genet Med. 2017;19(9):967-974.
ASMD diagnostic testing is a straightforward process, and may get patients the answers they need to begin managing their symptoms appropriately. Over time, ASMD symptoms can progress and lead to increased disease burden and risk of death.1,2
|
Adapted from McGovern MM et al. Genet Med. 2017;19(9):967-974.
aLimitations to DBS testing include the potential effects of anemia and recent transfusions on results. Skin fibroblasts can be used in equivocal cases.
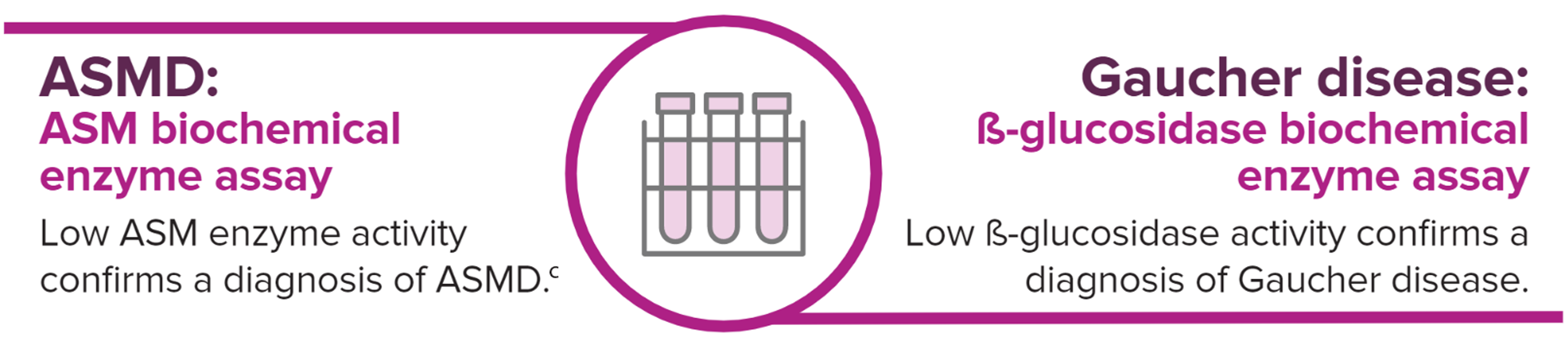
bGuidelines are based on a consensus of opinion from an international group of experts in ASMD.
cGenetic testing can also be performed as additional diagnostic confirmation for ASMD. However, gene sequencing should not be a substitute for the biochemical enzyme assay.
Have a question or want additional information? A Sanofi representative is available to answer your disease- or product-related questions. Click the link below to complete a request.
|
Symptoms & Diagnosis |
Monitoring |
For Hepatologists |
For Pulmonologists |
Sanofi's Medical Information Department can provide information on diagnostic testing, pharmacovigilance/safety, and ASMD.
|
Visit our Medical Information Website Please call 8 AM until 6 PM EST, Monday through Friday: 1-800-745-4447, option 2 (toll-free)/1-617-768-9000, option 2 |
CareConnect is a free, voluntary and confidential support program for eligible patients and families living with certain lysosomal storage disorders (LSDs).
Connected Education: Comprehensive disease education from diagnosis and beyond for individuals, families, and communities.
Connected Team: Experts who connect the dots between specialists, insurance, and appointments for a less fragmented care experience.
Connected Experience: Programs designed to support patients by connecting them with experts and the community to navigate life transitions and manage treatment.
If affording treatment is an issue, CareConnect may be able to help eligible patients access financial assistance. To learn more about our range of support offerings, connect with us at careconnectpss.com/hcp, call 1-800-745-4447, option 3, or email info@careconnectpss.com
Sanofi is committed to providing you with materials to help educate your patients about lysosomal storage disorders. A variety of patient educational materials are available to help patients understand ASMD, its symptoms, and management.

References: 1. McGovern MM et al. Orphanet J Rare Dis. 2017;12(1):41. 2. McGovern MM, et al. Genet Med. 2017;19(9):967-974. 3. McGovern MM et al. Pediatrics. 2008;122(2):e341-e349. 4. Cox GF et al. JIMD Rep. 2018;41:119-129. 5. Eyester KM. Adv Physiol Educ. 2007;31(1):5-16. 6. Schuchman EH et al. Mol Genet Metab. 2017;120(1-2):27-33. 7. McGovern MM et al. Neurology. 2006;66(2):228-232. 8. Cassiman D et al. Mol Genet Metab. 2016;118(3):200-213. 9. Kaplan P et al. Eur J Pediatr. 2013;172:447-458. 10. Mistry PK et al. Am J Hematol. 2011;86(1):110-115. 11. Grabowski GA et al. http://ommbid.mhmedical.com/content.aspx?bookid=474§ionid=45374148. Accessed March 2022. 12. Stirnemann J et al. Int J Mol Sci. 2017;18(2):441. 13. De Fost M et al. Atherosclerosis. 2009;204(1):267-272. 14. McGovern MM et al. Orphanet J Rare Dis. 2021;16(212):1-14. 15. Leukemia & Lymphoma Society. https://www.lls.org/leukemia/acute-lymphoblastic-leukemia/signs-and-symptoms. July 23, 2025. 16. Larson RA, Anastasi J. Acute lymphoblastic leukemia: clinical presentation, diagnosis, and classification. In: Estey EH, Faderl SH, Kantarjian HM, eds. Hematologic Malignancies: Acute Leukemias. Springer-Verlag; 2008:109-118. Hematologic Malignancies: Acute Leukemias. 2008. 17. Leukemia & Lymphoma Society. https://www.lls.org/lymphoma/non-hodgkin-lymphoma/signs-and-symptoms. Accessed March 2022. 18. American Cancer Society. https://www.cancer.org/cancer/non-hodgkin-lymphoma/treating/palliative-care.html. Accessed March 2022. 19. Shankland KR et al. Lancet. 2012;380(9844):848-857. 20. National Cancer Institute, National Institutes of Health. https://www.cancer.gov/types/lymphoma/patient/adultnhl-treatment-pdq. Accessed March 2022. 21. Liang TJ. Hepatology. 2009;49(5 suppl):S13-S21. 22. Watson RDS et al. BMJ. 2000;320(7229):236-239. 23. Kobelska-Dubiel N et al. Prz Gastroenterol. 2014;9(3):136-141.
MAT-US-2011636-v3.0-08/2025
