
Mucopolysaccharidosis type I (MPS I) clinical features and symptom presentation
Learn more
This website contains promotional content and is intended for Healthcare Professionals based in the United States only.
Understand the broad spectrum of clinical presentation
Mucopolysaccharidosis type I (MPS I) varies in the age of onset, symptom severity, progression rate, and presence of neurologic involvement.1,2
Timely intervention is key
Disease progression can lead to cardiovascular complications and respiratory disease, the leading morbidities associated with early death in MPS I.1,3
Long-term management and monitoring is essential
As a chronic, progressive, multisystemic disease, early multidisciplinary management is the best approach to slow progression.1,3
MPS I is a rare, genetic, debilitating, multisystemic, lysosomal storage disease. It is caused by the deficiency of lysosomal enzyme α-L-iduronidase (IDUA), which results in the progressive accumulation of nondegraded material (called glycosaminoglycans or GAGs) in cells throughout the body. The accumulated GAGs can cause cardiovascular complications and respiratory disease, which are the leading morbidities associated with early death in patients with MPS I.4,5
.jpg)
MPS I is caused by pathogenic variants in the gene that codes for lysosomal enzyme α-L-iduronidase (IDUA). MPS I is inherited in an autosomal recessive pattern and affects males and females equally. As MPS I is an inherited disease, this condition may affect other family members.4,5
Two common IDUA alleles, W402X and Q70X, and a minor IDUA allele, P533R, account for >50% of the MPS I alleles in the Caucasian demographic.4,5 To date, more than 200 pathogenic variants in the IDUA gene have been identified in patients with MPS I. Some variants result in more severe disease manifestations.6

GAG, glycosaminoglycan; IDUA, α-L-iduronidase
The IDUA enzyme is deficient in MPS I, resulting in GAG accumulation in cells which can cause tissue damage. Such damage can be irreversible and lead to loss of function, clinical deterioration, a variety of manifestations, and, in some cases, early death.2

In MPS I, the IDUA enzyme is deficient or essentially absent, causing GAG substratesa to accumulate in cells, leading to progressive damage to tissues and organs.2,4,7-11
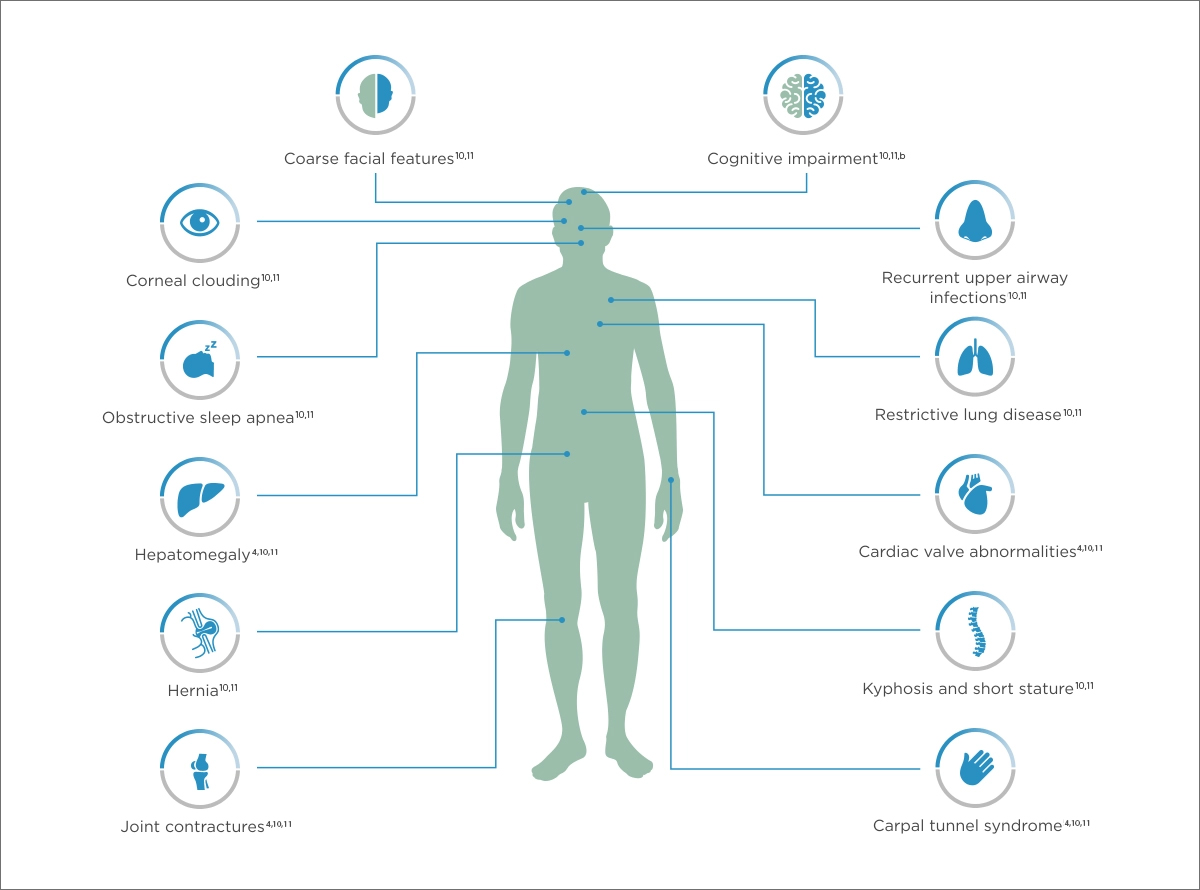
aThe GAGs that accumulate in MPS I are dermatan sulfate and heparan sulfate.7
bScheie form has no cognitive impairment.4
Cardiovascular manifestations |
Musculoskeletal manifestations |
|
|
Developmental delays |
Physical appearance |
|
|
This is not meant to be a comprehensive list of all manifestations associated with MPS I.
It is important to identify the common manifestations across the spectrum of MPS I to piece together the diagnosis of this potentially fatal disease.2
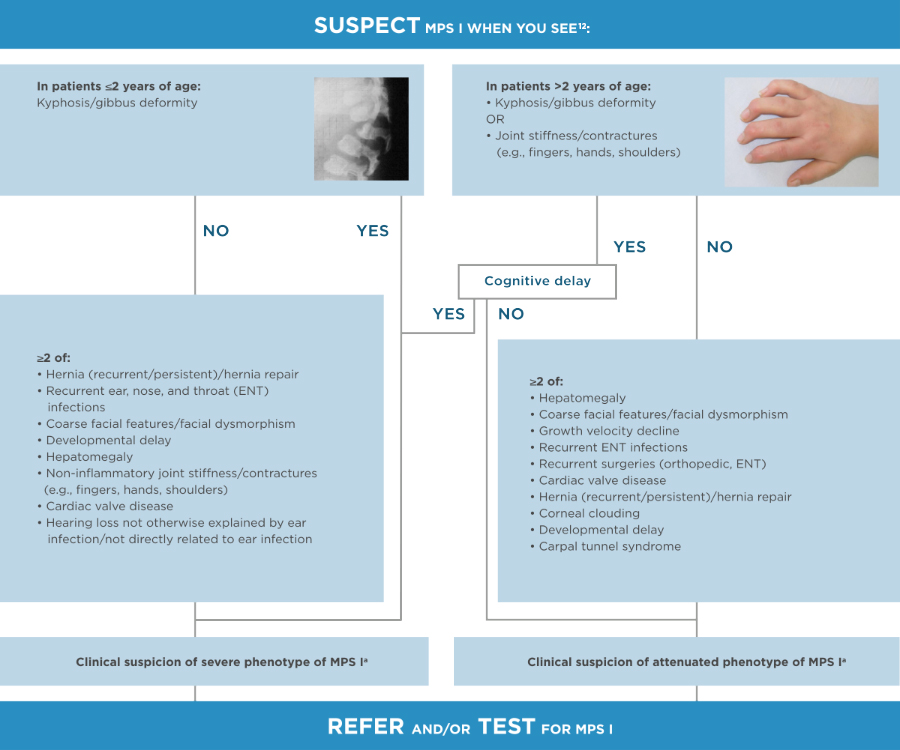
aUrinary GAGs testing to be done if available but without delaying the referral.
.2025-07-16-14-20-05.png)
|
|
|
The clinical success of HSCT depends on the age of the child at transplantation, the degree of clinical involvement, the child’s cardiopulmonary status and neurologic development, the type of donor, and the ability to achieve stable engraftment without the development of graft-versus-host disease.
Viral infections, graft rejection, pulmonary hemorrhage, and graft-versus-host disease (GvHD) remain the most common causes of death and are most commonly seen in the first several months after transplant, but can occur within the first year post-transplantation. These complications may be related to the immunologic responses of the recipient to the donor cells or vice versa, or may be due to the conditioning chemotherapy regimen, with use of agents such as busulfan and cyclophosphamide.15
This type of treatment helps in the management of symptoms. Examples of supportive care include:
ERT substitutes the naturally occurring enzyme that is deficient or absent in MPS I patients.
Early diagnosis is critical to the early initiation of disease management.17
The Recommended Schedule of Assessments represents the core MPS I disease-related assessments that allow for evaluations of disease progression over time. Physicians will determine the actual frequency of necessary assessments according to a patient’s individualized need for medical care and routine follow-up.17
| Initial Assessments | Every 6 Months | Every 12 Months | Every Other Year | |
| General | ||||
| Demographics | ■ | |||
| Patient Diagnosisa | ■ | |||
| Medical History | ■ | ■ | ||
| Physical Examination | ■ | ■ | ||
| General Appearance | ■ | ■ | ||
| MPS I Disease Clinical Assessments | ||||
| Neurologic/CNS | ||||
| ■ | ■ | ||
| ■ | ■ | ||
| ■ | ■ | ||
| Cognitive Testing (DQ/IQ) | ■ | ■ | ||
| Ophthalmologic | ||||
| ■ | ■ | ||
| ■ | ■ | ||
| ■ | ■ | ||
| Auditory | ||||
| ■ | ■ | ||
| Cardiac | ||||
| ■ | ■ | ||
| ■ | ■ | ||
| Respiratoryb | ||||
| ■ | ■ | ||
| ■ | ■ | ||
| Gastrointestinal | ||||
| ■ | ■ | ||
| ■ | ■ | ||
| Musculoskeletal | ||||
| ■ | ■ | ||
| Vitals and Laboratory Tests | ||||
| ■ | ■ | ||
| ■ | ■ | ||
| ■ | ■ | ||
| ■ | |||
| ■ | ■ | ||
| ■ | ■ | ||
| Functional Outcome Measurements | ||||
| ■ |
All tests requiring sedation are recommended only if sedation is considered to be safe for the patient.
aDiagnosis would include genotype, if available.
bMay not be possible in non-cooperative patients or patients younger than 5 to 6 years of age.
cThe recommended method for obtaining organ volumes is MRI or computed tomography to enable quantitative analysis. However, if in the opinion of the clinician it is unsafe to sedate the patient, then ultrasound may be substituted.
dMeasured in pediatric patients only, unless determined otherwise by treating physician.
CNS, central nervous system; DQ, developmental quotients; ECG, electrocardiogram; FVC, forced vital capacity; FEV, forced expiratory volume; GAGs, glycosaminoglycans; IQ, intelligence quotient; MPS, mucopolysaccharidosis
Have a question or want additional information? A Sanofi representative is available to answer your disease- or product-related questions. Click the link below to complete a request.
Sanofi’s Medical Information Department can provide information on diagnostic testing, pharmacovigilance/safety, and MPS I disease.
|
Visit our Medical Information Website Please call 8 AM until 6 PM EST, Monday through Friday: 1-800-745-4447, option 2 (toll-free)/1-617-768-9000, option 2 |
CareConnect is a free, voluntary and confidential support program for eligible patients and families living with certain lysosomal storage disorders (LSDs).
Connected Education: Comprehensive disease education from diagnosis and beyond for individuals, families, and communities.
Connected Team: Experts who connect the dots between specialists, insurance, and appointments for a less fragmented care experience.
Connected Experience: Programs designed to support patients by connecting them with experts and the community to navigate life transitions and manage treatment.
If affording treatment is an issue, CareConnect may be able to help eligible patients access financial assistance. To learn more about our range of support offerings, connect with us careconnectpss.com/hcp, call 1-800-745-4447 option 3, or email info@careconnectpss.com
This listing is provided as a resource only and does not constitute an endorsement by Sanofi of any particular organization or its programming. Additional resources on this topic may be available and should be investigated. Sanofi does not review or control the content of non-Sanofi websites.
Resources for your patients:
The following professional organizations are dedicated to genetic diseases:


References: 1. Clarke LA. GeneReviews® [Internet]. Accessed April 11, 2025. https://www.ncbi.nlm.nih.gov/books/NBK1162/ 2. Beck M et al. Genet Med. 2014;16(10):759-765. 3. Muenzer J et al. Pediatrics. 2009;123(1):19-29. 4. Neufeld EF et al. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA, eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill; 2019. Accessed March 21, 2022. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225069235 5. Vijay S, Wraith JE. Acta Paediatr. 2005;94(7):872-877. 6. Poletto E et al. Clin Genet. 2018;94(1):95-102. 7. de Ru MH et al. Orphanet J Rare Dis. 2011;6:55. 8. Wraith JE et al. J Pediatr. 2004;144(5):581-588. 9. D’Aco K et al. Eur J Pediatr. 2012;171(6):911-919. 10. Cleary MA, Wraith JE. Acta Paediatr. 1995;84(3):337-339. 11. Scheie HG et al. Am J Ophthalmol. 1962;53:753-769. 12. Tylki-Szymańska A et al. Acta Paediatr. 2018;107(8):1402-1408. 13. Wraith JE. Expert Opin Pharmacother. 2005;6(3):489-506. 14. Taylor M et al. Biol Blood Marrow Transplant. 2019;25(7):e226-e246. 15. Hampe CS et al. Biomolecules. 2021;11(2):189. 16. Nan H et al. Biomed Res Int. 2020;2020:2408402. 17. Pastores GM et al. Mol Genet Metab. 2007;91(1):37-47. 18. Berger KI et al. J Inherit Metab Dis. 2012;36(2):201-210. 19. Braunlin EA et al. J Inherit Metab Dis. 2011;34(6):1183-97. 20. White KK. Rheumatology. 2011;50(Suppl5):v26-v33. 21. Muenzer J et al. Pediatrics. 2009;123:19-29.
MAT-US-2205119-v2.0-08/2025
