- Article
- Source: Campus Sanofi
- Feb 27, 2026
Mucopolysaccharidosis type I (MPS I) clinical features and symptom presentation
Patients with mucopolysaccharidosis (MPS) I face a multitude of serious and often debilitating multisystemic manifestations.
In MPS I, the alpha-L-iduronidase (IDUA) enzyme is deficient or essentially absent, causing glycosaminoglycans (GAGs) substrates to accumulate in cells, leading to progressive damage to tissues and organs.1-7 Learn more about the pathophysiology of MPS I.
Understanding the range of mucopolysaccharidosis type I symptoms
MPS I can present with a wide spectrum of signs and symptoms. It is therefore important to identify the common manifestations across the spectrum of MPS I to piece together the diagnosis of this potentially fatal disease.6
MPS I characteristics
The types of MPS I include the severe form (Hurler syndrome) and the attenuated form (Hurler-Scheie and Scheie syndrome). These phenotypes differ in disease severity and speed of progression, however, show overlapping clinical features.6,8
Hurler and Hurler-Scheie syndrome symptoms are both characterized by cognitive deficits8 while Scheie syndrome has no cognitive impairment.7
MPS I Can Present with a Wide Array of Signs and Symptoms



Features and symptoms of severe and attenuated MPS I forms
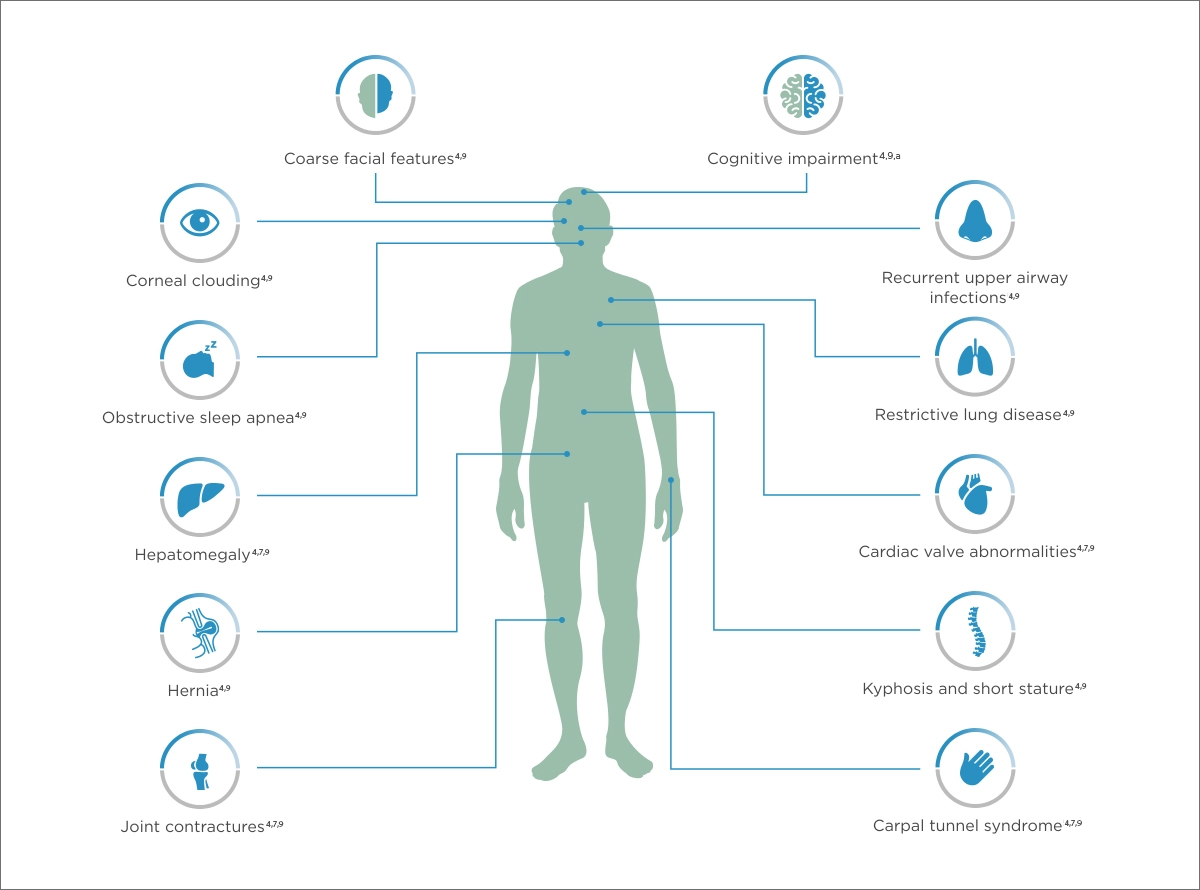
aScheie form has no cognitive impairment.
The MPS I disease spectrum is marked by heterogeneity that can complicate diagnosis, with varying ages of onset, unusual symptom combinations, and a broad range of sometimes life-limiting and life-threatening clinical manifestations.6,8
Mucopolysaccharidosis type I facial features
The appearance of some infants with severe and moderate-to-severe symptoms of MPS I may appear unaffected at birth, but as early as 3 months of age, parents may notice changes in their child’s facial features.9
Pronounced facial features of MPS I include:
- Enlarged lips and cheeks5
- Short noses, flat faces, prominent foreheads4
- Large heads, which tend to be longer than normal from front to back (scaphocephaly)4
Some individuals with the severe form of MPS I have macroglossia, characterized by an abnormally large tongue which protrudes past the dental arch when the mouth is closed, making swallowing and speaking difficult.10,11
Mucopolysaccharidosis type I ocular manifestations
Corneal clouding of the eye is usually one of the first ocular manifestations in patients with the severe form of MPS I.11–13
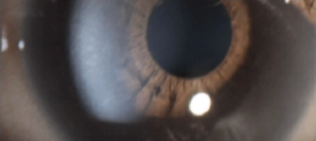
The accumulation of GAGs and subsequent disorganization of collagen fibrils in the cornea cause corneal opacity.11,13 Corneal clouding often starts within the first year of life, causing vision impairment and often leading to blindness.12,14 Retinal degeneration and optic nerve compression can also manifest in children.12,13
Mucopolysaccharidosis type I musculoskeletal manifestations
Adults are often stocky in build, their trunks shorter than their limbs, and their necks may be short and stiff.4,9 Bone abnormalities are common in those with MPS I, including:
- Hip deformations15
- Ovoid vertebrae15
- Widening of the ribs.15
- Poorly formed pelvis7,15
- Gibbus deformity of the back7,15
- Shortened phalanges of the hands7,15
- Joint contractures7,16
.2025-07-07-12-50-40.png)
Skeletal dysplasia can occur in the severe types of MPS I but is frequently missing or subclinical in patients with attenuated MPS I.15
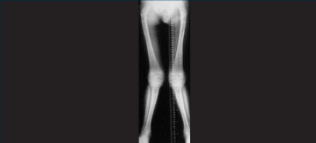
Joint stiffness, pain and contractures are common among patients with attenuated MPS I.7 Evolving joint pain and joint contractures in the absence of inflammation should always raise suspicion of an MPS disorder.16

Limited range of motion mobility in the absence of inflammation should raise suspicion of MPS I.
Cardiovascular manifestations
Cardiovascular manifestations are common in patients with MPS I.18 In particular, cardiomyopathy and endocardial fibroelastosis can occur in children with severe MPS I.11 Patients with an attenuated form of MPS I may develop aortic or mitral valve disease, and this can lead to a slowly progressive valvular heart disease over several years.18 In some cases, these patients may need mitral or aortic valve replacement as early as in their teens and twenties.18
Developmental manifestations
Developmental delays are apparent in the severe form of MPS I starting at 12–24 months of age, typically with a maximum functional age of 2–4 years.19 The initial presentation of delay is then followed by slowed development and progressive regression in developmental skills until death.20
Mildly affected patients with the attenuated form generally have unaffected cognition.7
Diagnosing and managing mucopolysaccharidosis type I patients
Recognizing symptoms and making an accurate early diagnosis are critical for MPS I disease management.3 Some common symptoms of MPS I can be similar to the symptoms of other diseases.
| MPS I 21 | Juvenile rheumatoid arthritis22 | Progressive pseudo- rheumatoid Dysplasia23 | Hypothyroidism 24 | Skeletal dysplasia 25 | Other MPS types26 | |
| Growth Retardation | ■ | ■ | ■ | ■ | ■ | |
| Joint Contractures Disease | ■ | ■ | ■ | ■ | ■ | |
| Skeletal Manifestations | ■ | ■ | ■ | ■ | ||
| Cardiac Disease | ■ | ■ | ■ | ■ | ||
| Hernia | ■ | ■ | ■ | |||
| Respiratory Disease | ■ | ■ | ■ |
As MPS I can affect multiple organ systems, diagnosis and treatment may require collaboration and communication among geneticists, neurologists, pediatricians, developmental specialists, surgeons, cardiologists, gastroenterologists, physical therapists, and primary care providers.3
References: 1. de Ru MH et al. Orphanet J Rare Dis. 2011;6(55):1-9. 2. Wraith JE. Expert Opin Pharmacother. 2005;6(3):489-506. 3. D’Aco K et al. Eur J Pediatr. 2012;171(6):911-919. 4. Scheie HG et al. Barness LA. Am J Ophthalmol. 1962;53:753-769. 5. Hampe CS et al. Cells. 2020;9(8):1838. 6. Beck M et al. Genet Med. 2014;16(10):759-765. 7. Neufeld EF et al. The Online Metabolic and Molecular Bases of Inherited Disease. Accessed May 1, 2025. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225544161 8. Kubaski F et al. Diagnostics (Basel). 2020;10(3):161. 9. Cleary MA, Wraith JE. Acta Paediatr. 1995;84(3):337-339. 10. Kutti Sridharan G, Rokkam VR. In: StatPearls. StatPearls Publishing;2025. Accessed May 1, 2025. https://www.ncbi.nlm.nih.gov/books/NBK560545/ 11. Sakuru R, Bollu PC. In: StatPearls. StatPearls Publishing;2025. Accessed May 1, 2025. https://www.ncbi.nlm.nih.gov/books/NBK2261/ 12. Ashworth JL et al. EYE. 2006;20(5):553-563. 13. Tomatsu S et al. J Clin Med. 2019;8(9):1467. 14. Vance M et al. Sci Rep. 2016;6(1):22131. 15. Clarke LA. GeneReviews® [Internet]. Accessed May 1, 2025. https://www.ncbi.nlm.nih.gov/books/NBK1162/ 16. Cimaz R et al. Pediatr Rheumatol Online J. 2009;7(1):18. 17. Guffon N et al. Eur J Pediatr. 2019;178(4):593-603. 18. Braunlin EA et al. J Inherit Metab Dis. 2011;34(6):1183-1197. 19. Shapiro EG et al. Mol Genet Metab. 2017;122S:1-7. 20. Shapiro EG et al. Mol Genet Metab. 2015;116(1-2):61-68. 21. Tylki-Szymanska A et al. Acta Paediatr. 2018;107(8):1402-1408. 22. Thatayatikom A et al. StatPearls [Internet]. StatPearls Publishing; 2022. Accessed August 17, 2025. https://www.ncbi.nlm.mih.gov/books/BNK554605/ 23. Bhavani GSL et al. In: Adam MP, Arranger HH, Pagon RA, eds. GeneReviews® [Internet]/ University of Washington, Seattle; 1993-2022. Accessed August 17, 2025. 24. Gupta V et al. Indian J Endocrinol Metab. 2011;15(Suppl 2):S99-S106. 25. Krakow D. Clin Perinatol. 2015;41(2):301-319, viii. 26. Lorne C et al. J Inborn Errors Metab Screen. 2018;7:1-12.
MAT-US-2509585-v1.0-02/2026
