- Wissen
- Quelle: Campus Sanofi
- 08.02.2026
Morbus Fabry - Therapie

Therapeutische Ansätze bei Morbus Fabry
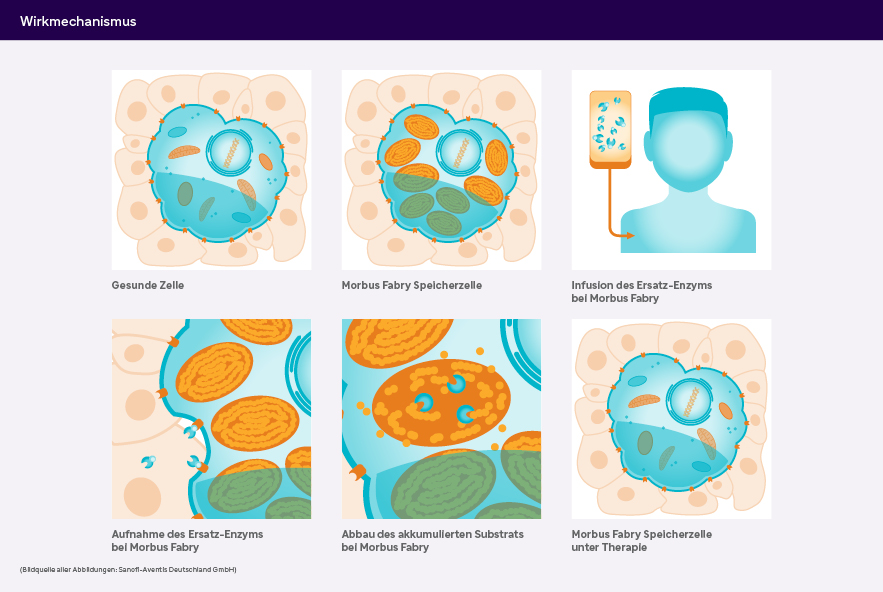
Bei Morbus Fabry kommt es aufgrund eines defekten Gens zu einem Mangel an α-Galaktosidase A. Das führt zu einer kontinuierlichen Speicherung von Glykosphingolipiden (Globotriaosylceramid, GL-3) in den Lysosomen des vaskulären Endothels und verschiedener Gewebe. Da es sich um eine genetisch bedingte, progredient verlaufende Erkrankung handelt, muss die Therapie bei Morbus Fabry ein Leben lang erfolgen.1,2
Enzymersatztherapie bei Morbus Fabry und ihre Vorteile
Seit 2001 sind zwei spezifische Enzymersatztherapien mit einer biotechnologisch hergestellten Form der humanen α-Galaktosidase A verfügbar. Das rekombinante Enzym wird regelmäßig i.v. infundiert, um den Mangel an endogenem Enzym auszugleichen. Über den Blutkreislauf erreicht es die Zielzellen, wo es mit Hilfe von Mannose-6-Phosphat-Rezeptoren durch die Zellmembran in die Lysosomen transportiert wird. Dort sorgt es für den Abbau der akkumulierten Glykosphingolipide.3 Seit 2023 ist noch eine weitere Form der Enzymersatztherapie zugelassen.
Eine weitverbreitete Zelllinie zur Herstellung einer Enzymersatztherapie ist die CHO-Zelle.
Erfahren Sie im folgenden Video, wie aus diesen Zellen ein Medikament entsteht.
Die Chaperontherapie bei Morbus Fabry
Pharmakologisch wirksame Chaperone sollen durch Beeinflussung der fehlerhaften Proteinkonformation deren Restenzymaktivität erhöhen. Sie lagern sich dabei spezifisch an die Zielproteine an, begleiten sie an den Zielort, die Lysosomen, und lösen sich dann wieder von diesen ab. Die Chaperontherapie greift nur bei jenen krankheitsauslösenden Mutationen, die zu einem fehlgefalteten Protein führen. Bei Mutationen, die beispielsweise zu deutlich verkürzten Proteinen führen oder bei denen das Protein komplett fehlt, funktioniert diese Therapieform naturgemäß nicht (non-amenable mutation). Seit 2016 ist eine Chaperontherapie zur Behandlung des Morbus Fabry für Patienten ab 12 Jahre verfügbar, die eine zugängliche Mutation haben und die noch eine ausreichende Nierenrestfunktion besitzen (eGFR ≥30 ml/min/1,73 m2).
Weitere Behandlungsmöglichkeiten bei Morbus Fabry
Abhängig von der individuellen Ausprägung der Symptomatik können zusätzliche Therapien erforderlich werden:
| Adjuvante Therapie bei Morbus Fabry | |
Symptomatische Begleittherapie1 |
Anpassung der Lebensgewohnheiten an die Erkrankung:
|
Medizintechnische Begleittherapie1 |
|
|
Schmerztherapie4
|
|
Therapiemonitoring
Im Rahmen des Langzeittherapiemonitoring sollten alle betroffenen Organsysteme regelmäßig sorgfältig überwacht und vor allem auch die Herz- und Nierenfunktion engmaschig kontrolliert werden. Der Biomarker Lyso-GL-3 kann Aufschluss über das Ansprechen auf die Enzymersatztherapie liefern.
Organfunktionen
Im Rahmen des Therapiemonitorings sollten insbesondere Niere, Herz und neurologische Parameter regelmäßig erfasst und überwacht werden.
Biomarker
Zusätzlich hat sich das lösliche Lyso-GL-3 (die deacetylierte Form von GL-3) im Plasma als zuverlässiger Biomarker für die Verlaufskontrolle bewährt.
Fabry-Zentren
Die Einleitung der Therapie und regelmäßige, interdisziplinäre Kontrolluntersuchungen können in einem der auf M. Fabry spezialisierten Zentren erfolgen. Den Kontakt zu einem Expertenzentrum in Ihrer Nähe vermitteln wir gerne auf Anfrage.
Erfahren Sie mehr zu den Kompetenzzentren:
Ernährungsempfehlungen
Grundsätzlich kann eine gesunde und ausgewogene Ernährung das Wohlbefinden steigern. Auch Patient*innen mit Morbus Fabry sollten:
- bei Übelkeit oder Krämpfen lieber häufiger kleinere Mahlzeiten zu sich nehmen und auf zu fette Nahrungsmittel verzichten
- bei Bluthochdruck oder einer kardialen Beteiligung auf eine salzarme Ernährung achten
- bei Nierenerkrankungen in Rücksprache mit ihrem Arzt/ihrer Ärtzin auf eine proteinarme Ernährung achten. Manche Patient*innen entwickeln Probleme hinsichtlich des Kaliumspiegels – hier ist es wichtig, auch diesen im Blick zu halten und auf kaliumarme Lebensmittel zu verzichten.


Mehr zur Morbus Fabry Therapie finden Sie auch im Login Bereich:
Erfahren Sie mehr über Morbus Fabry:
Häufig gestellte Fragen zur Therapie von Morbus Fabry
Welche Therapieoptionen gibt es bei Morbus Fabry?
Bei Morbus Fabry stehen verschiedene Therapien zur Verfügung. Die primären Therapieansätze sind die Enzymersatztherapie und die Chaperontherapie.
Seit 2001 sind zwei spezifische Enzymersatztherapien mit biotechnologisch hergestellter α-Galaktosidase A verfügbar, die regelmäßig intravenös infundiert wird.3 Eine weitere Form der Enzymersatztherapie ist seit 2023 zugelassen. Die Chaperontherapie ist seit 2016 für Patient*innen ab 12 Jahren mit zugänglicher Mutation und ausreichender Restenzymaktivität verfügbar. Da Morbus Fabry eine genetisch bedingte, progredient verlaufende Erkrankung ist, muss die Therapie ein Leben lang erfolgen.1, 2
Welche begleitenden Behandlungen sind bei Morbus Fabry wichtig?
Abhängig von der individuellen Symptomatik können zusätzliche Therapien erforderlich sein. Dazu gehören die Anpassung der Lebensgewohnheiten (bspw. Vermeidung von Hitze, Stress, körperlicher Anstrengung), spezielle Bewegungsprogramme, individuell abgestimmte Ernährung und psychosoziale Betreuung.
Bei fortgeschrittener Erkrankung können medizintechnische Maßnahmen wie Dialyse, Nierentransplantation oder Herzschrittmacher-Implantation notwendig werden. Zur Schmerztherapie werden häufig NSAR, Antikonvulsiva, Antidepressiva oder Opioide eingesetzt, deren Art und Dosierung individuell angepasst werden müssen.1, 4
Wie wird der Therapieerfolg bei Morbus Fabry überwacht?
Im Rahmen des Langzeittherapie-Monitorings sollten alle betroffenen Organsysteme regelmäßig überwacht werden, besonders die Herz- und Nierenfunktion. Das lösliche Lyso-GL-3 im Plasma hat sich als zuverlässiger Biomarker für die Verlaufskontrolle bewährt. Die Einleitung der Therapie und regelmäßige interdisziplinäre Kontrolluntersuchungen sollten in einem der auf Morbus Fabry spezialisierten Zentren erfolgen.
- Germain DP. Orphanet J Rare Dis 2010; 5:30.
- Desnick RJ et al. α-Galactosidase A deficiency: Fabry disease. In: C. R. Scriver, A. L. Beaudet, W. S. Sly, D. Valle (Hrsg.): The metabolic and molecular basis of inherited disease. 8. Ausgabe, Verlag McGraw-Hill, 2001,S.3733–3774.
- Migeon BR. J Am Soc Nephrol 2008; 19: 2052-9.
- Fachinformation Fabrazyme, Stand 03/2024.
Header-Foto: © iStock (Jacob Wackerhausen) MAT-DE-2202170 - v3.0 - 02/2026
