- Artikel
- Bron: Campus Sanofi
- 16 apr 2025
Understanding Gaucher Disease: Symptoms, Classification, and Clinical Presentation

About Gaucher Disease
Underlying pathology
The disease was first described by French medical student Phillipe Gaucher in 1882, when he observed a young woman with an enlarged spleen and characteristic engorged cells.2 A little more than 50 years later, Aghion reported that people with this disease accumulated a sphingolipid called glucosylceramide. But it wasn't until 1965 that Brady and colleagues showed that Gaucher disease was caused by reduced activity of the enzyme acid β-glucosidase.2
The functional enzyme has the activity of decomposing glucosphingolipids derived from the physiological renewal of membranes, particularly blood cells. Pathogenic variants in the acid β-glucosidase gene trigger a reduction in the enzyme activity of this enzyme, which then becomes insufficient to prevent the accumulation of a glucosphingolipid, called glucosylceramide, in the lysosomes of cells, mainly in macrophages. Macrophages with accumulated glucosylceramide, called Gaucher cells, accumulate in the organs. The storage of Gaucher cells triggers a cascade of pathophysiological events, including the production of a chronic and hypermetabolic inflammatory state.3
This is a multisystem disease that has significant variations in its clinical manifestations, severity and course. A partial deficiency of acid β-glucosidase is associated with diseases of the liver, spleen, bone marrow, and lung. A severe deficiency of the enzyme is also associated with neurological manifestations.4
Gaucher cell by accumulation of glucosylceramide (and other substrates)
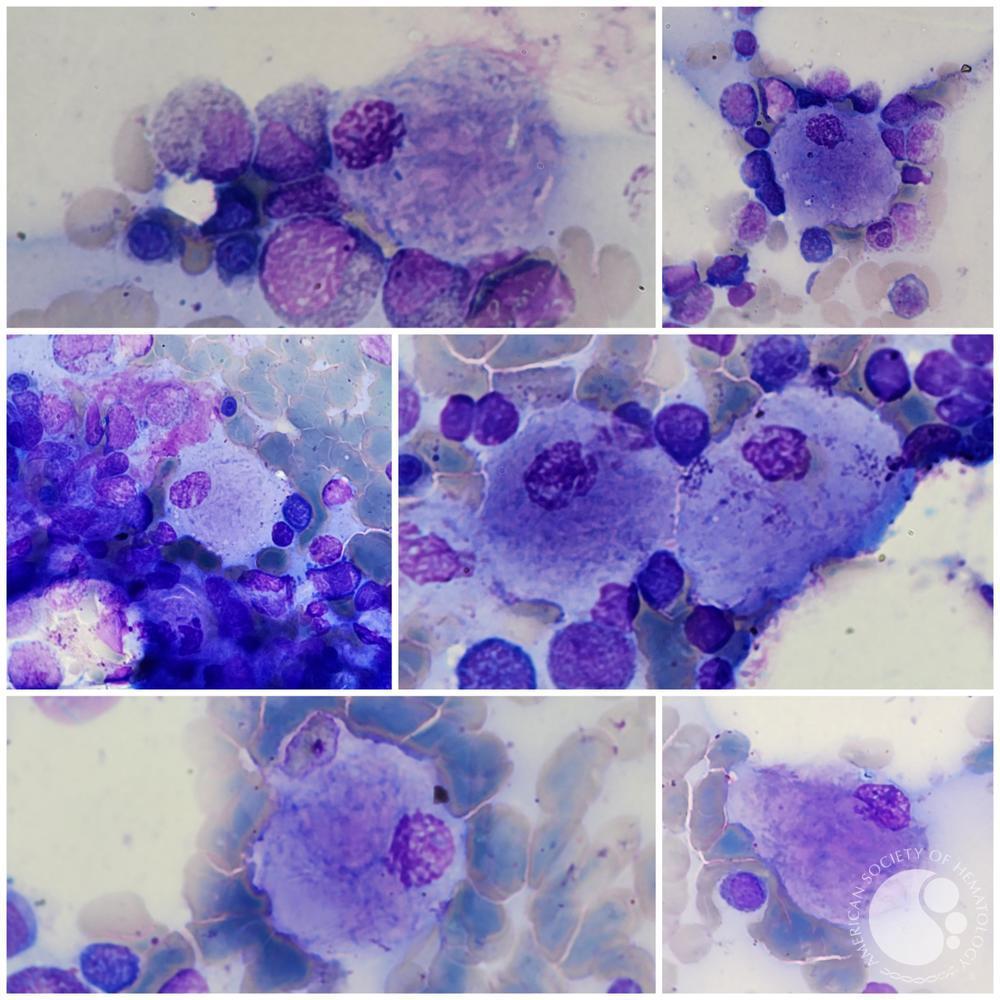
Different types of gaucher cells with an appearance of "crumpled paper" can be seen in the bone marrow aspiration of a patient with Gaucher disease.5
Classification of the disease
Gaucher disease is generally classified into three types, based on the absence (type 1) or presence (types 2 and 3) of neurological involvement1:
Type 1 (non-neuropathic)
Type 1 (non-neuropathic) has a wide variation in its clinical manifestations, severity and course, but is distinguished from the other two types by the absence of central nervous system involvement.7 In some people, symptoms begin in early childhood and worsen over time, while in others, the first symptoms may not be noticed until adulthood.6
Type 2 (acute neuropathic)
Type 2 (acute neuropathic) is usually evident in the first few months of life and includes severe neurological symptoms. These children usually do not survive from the age of 2.8
Type 3 (chronic neuropathic)
Type 3 (chronic neuropathic) is characterized by a slowly evolving neurological disease, that can have visceral and hematologic signs like in type 1 disease in its early stages. Patients who reach adolescence can live to adulthood.6
Type 1 (non-neuropathic)
Type 1 (non-neuropathic) has a wide variation in its clinical manifestations, severity and course, but is distinguished from the other two types by the absence of central nervous system involvement.7 In some people, symptoms begin in early childhood and worsen over time, while in others, the first symptoms may not be noticed until adulthood.6
Type 2 (acute neuropathic)
Type 2 (acute neuropathic) is usually evident in the first few months of life and includes severe neurological symptoms. These children usually do not survive from the age of 2.8
Type 3 (chronic neuropathic)
Type 3 (chronic neuropathic) is characterized by a slowly evolving neurological disease, that can have visceral and hematologic signs like in type 1 disease in its early stages. Patients who reach adolescence can live to adulthood.6
Categorization of the different types of Gaucher disease is valuable in terms of assessing disease management options and as a basis for genetic counseling. The disease is described as a continuum phenotype, with a spectrum of symptoms ranging from mild to severe neurological effects.8
Clinical presentation
Gaucher disease is a multisystem disease with great phenotypic variation between patients. The heterogeneous symptoms observed often make diagnosis a significant challenge. However, there are some symptoms and signs that are more frequently observed in the different types of Gaucher disease.10
Splenomegaly is one of the most common symptoms in type 1 Gaucher disease and can often be the first finding to be recognized when a child is young.4 However, these patients may also develop skeletal diseases, possibly resulting in generalized osteoporosis complicated by frequent pathological fractures. Skeletal abnormalities are also very common in almost all adult patients who develop bone complications.4 There is a broad spectrum of severity of a number of signs and symptoms, including:10
Visceral
- Splenomegaly
- Hepatomegaly
Haematological
- Easy bruising and bleeding associated with thrombocytopenia
- Anaemia
Skeletal
- Bone crises/pain
- Growth retardation (children and adolescents)
- Avascular necrosis
- Pathological fractures
- Osteopenia
- Bone marrow infiltration
Quality of life
- Type 1 Gaucher disease is characterized by the absence of primary involvement of the central nervous system
Gaucher symptoms prevalence data from Charrow J et al.11
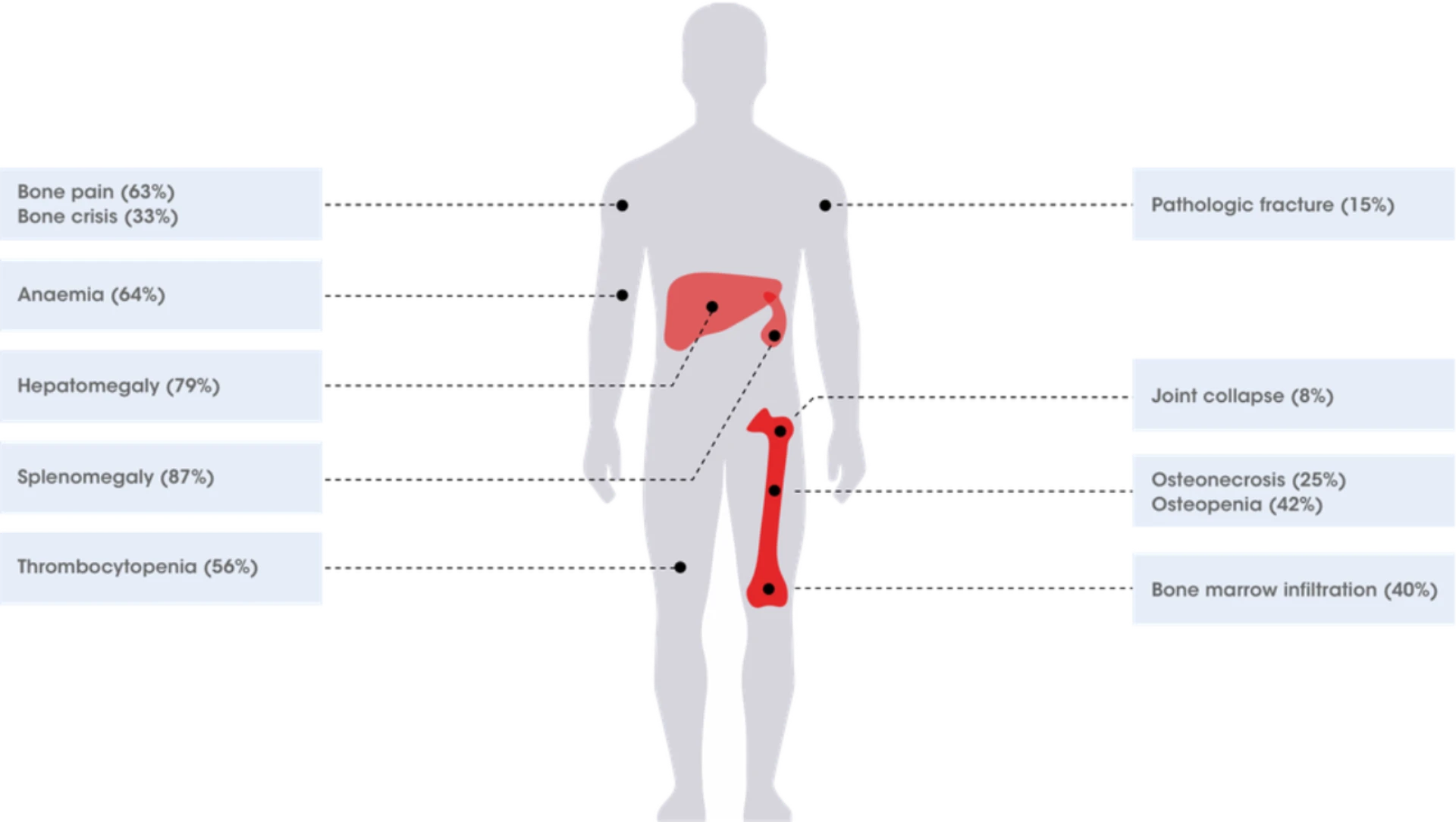
Splenomegaly – one of the most common signs of Gaucher disease.4, 11 A study from the Gaucher Registry described the clinical characteristics of 1698 Gaucher patients (94% type 1, 1% type 2 and 5% type 3) prior to receiving enzyme replacement therapy.11
Type 2 Gaucher disease also has a spectrum of phenotypes. This acute neuropathic type is the most severe form of the disease and includes a number of signs and symptoms associated with type 1 disease and a number of additional neurological manifestations that typically result in premature death. Characteristics of type 2 Gaucher disease include:8
- Supranuclear eye palsy
- Opisthotonus
- Hydrops fetalis
- Ichthyosis-like skin disorder
- Eye complications including strabismus
- Trismus
The severity of type 3 Gaucher disease (chronic neuropathic) is generally considered to be between types 1 and 2, although there are also less severe forms of type 3 Gaucher disease. As with type 1 disease, type 3 Gaucher disease is characterized by a series of visceral, hematological and bone manifestations, to which is added neurological involvement (with a later onset and less severity compared to type 2 disease):6
- Supranuclear eye palsy
- Ataxia6
Splenomegaly is one of the most common symptoms in type 1 Gaucher disease and can often be the first finding to be recognized when a child is young.4 However, these patients may also develop skeletal diseases, possibly resulting in generalized osteoporosis complicated by frequent pathological fractures. Skeletal abnormalities are also very common in almost all adult patients who develop bone complications.4 There is a broad spectrum of severity of a number of signs and symptoms, including:10
Visceral
- Splenomegaly
- Hepatomegaly
Haematological
- Easy bruising and bleeding associated with thrombocytopenia
- Anaemia
Skeletal
- Bone crises/pain
- Growth retardation (children and adolescents)
- Avascular necrosis
- Pathological fractures
- Osteopenia
- Bone marrow infiltration
Quality of life
- Type 1 Gaucher disease is characterized by the absence of primary involvement of the central nervous system
Gaucher symptoms prevalence data from Charrow J et al.11
Splenomegaly – one of the most common signs of Gaucher disease.4, 11 A study from the Gaucher Registry described the clinical characteristics of 1698 Gaucher patients (94% type 1, 1% type 2 and 5% type 3) prior to receiving enzyme replacement therapy.11
Type 2 Gaucher disease also has a spectrum of phenotypes. This acute neuropathic type is the most severe form of the disease and includes a number of signs and symptoms associated with type 1 disease and a number of additional neurological manifestations that typically result in premature death. Characteristics of type 2 Gaucher disease include:8
- Supranuclear eye palsy
- Opisthotonus
- Hydrops fetalis
- Ichthyosis-like skin disorder
- Eye complications including strabismus
- Trismus
The severity of type 3 Gaucher disease (chronic neuropathic) is generally considered to be between types 1 and 2, although there are also less severe forms of type 3 Gaucher disease. As with type 1 disease, type 3 Gaucher disease is characterized by a series of visceral, hematological and bone manifestations, to which is added neurological involvement (with a later onset and less severity compared to type 2 disease):6
- Supranuclear eye palsy
- Ataxia6
-
Mistry PK, Weinthal JA, Weinreb NJ. Disease state awareness in Gaucher disease: a Q&A expert roundtable discussion. Clin Adv Hematol Oncol. 2012;10:1-16.
-
Brady RO. Gaucher’s disease: past, present and future. Bailleres Clin Haematol 1997;10(4):621-34.
-
Grabowski G. Gaucher disease and other storage disorders. Hematology Am Soc Hematol Educ Program 2012;1:13-18 (doi: 10.1182/asheducation-2012.1.13)
-
Cox TM, Schofield JP. Gaucher’s disease: clinical features and natural history. Bailleres Clin Haematol 1997;10(4):657-689.
-
Ajmaldin Saki Ph.D; Bita Bandar M.Sc; Narjes Sadat Sadati M.Sc; Hakimeh Hadi M.Sc. https://imagebank.hematology.org/image/64378/gaucher-cells
-
Grabowski GA, Petsko GA, Kolodny EH. Gaucher Disease. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Education; 2019. Accessed October 06, 2025. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225546056
-
Kaplan P, Baris H, De Meirleir L, Di Rocco M, El-Beshlawy A, Huemer M, Martins AM, Nascu I, Rohrbach M, Steinbach L, Cohen IJ. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr (2013) 172:447–458
-
Gupta N, Oppenheim I, Kauvar E, Tayebi N, Sidransky E. Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity. Blood Cells Mol Dis 2011;46(1):75-84.
-
Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 2004;83(1-2):6-15.
-
Damiano AM, Pastores GM, Ware JE Jr. The health-related quality of life of adults with Gaucher disease receiving enzyme replacement therapy: results from a retrospective study. Qual Life Res 1998;7(5):373-386.
-
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, Weinreb NJ, Zimran A. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835.
Neem contact op

MAT-BE-2501471