- Artikel
- Bron: Campus Sanofi
- 2 mrt 2026
Gaucher disease in children

Classification and early presentation
Gaucher disease is an autosomal recessive lysosomal storage disorder caused by pathogenic variants in the GBA1 gene, resulting in deficient activity of the enzyme acid β glucosidase. This deficiency leads to accumulation of glucosylceramide and related lipids within macrophages, forming characteristic Gaucher cells.1-5 These lipid laden cells infiltrate multiple organs, particularly the spleen, liver, bone marrow and lungs.1,2
Why early recognition of Gaucher disease matters
Early recognition in paediatric patients enables timely intervention, preventing irreversible skeletal, hematologic, and visceral complications.1,2 Various types of treatment are available for patients with Gaucher disease, which focuses on addressing the underlying metabolic defect to reduce disease burden and improve organ function, alongside supportive care for hematologic, skeletal, and growth-related complications. Therapy is individualized based on disease severity, symptoms, and progression, with regular monitoring to guide initiation and adjustment of treatment strategies.1
Gaucher disease should be considered in children
- frequently presenting with splenomegaly which often emerges before other symptoms and sometimes as the only early sign4
- with hepatomegaly, thrombocytopenia, and anaemia if there is no alternative explanation2,3
- with bone manifestations, including bone pain, delayed growth, Erlenmeyer-flask deformity, and bone crises1,4
- with growth delay and delayed puberty2
Clinical types
Gaucher disease comprises three major clinical types ranging from neuronopathic to severe neuronopathic forms, of which type 1 is the most seen form in children (table 1).3,6-9
Type |
Neurologic involvement |
Typical onset |
Key features |
| Type 1 (non neuropathic) | None | Childhood to adulthood | Hepatosplenomegaly, cytopenias, bone disease |
| Type 2 (acute neuropathic) | Severe, early | Infancy | Rapid neurological decline, death in early childhood |
| Type 3 (chronic neuropathic) | Slowly progressive | Childhood | Neurologic impairment + systemic manifestations |
Table 1. Clinical types of Gaucher disease3,6-9
Clinical presentation in children
Children with Gaucher disease almost always (95%) present with splenomegaly, appearing earlier than bone manifestations. In half of young children, splenomegaly is the only initial symptom.1,4
Hepatomegaly (present in 87% of cases), thrombocytopenia (50%) and anaemia (40%) are among the most common presenting combinations in children with Gaucher disease.1,2,4
Gaucher disease should be considered when splenomegaly coexists with cytopenias or bone pain.1,4
Key early symptoms1,2,3
Visceral manifestations
Hematologic findings
|

|
Skeletal disease
Neurological features (Types 2 and 3)
|
Age at which symptoms may occur
Symptoms of Gaucher disease can begin in infancy, especially in the more severe forms like Gaucher disease type 2, which often presents prenatally, perinatally, or in the first few months of life, and typically leads to death in infancy. For Gaucher disease type 3, children often show symptoms within the first year and later in childhood, including neurological issues like seizures and saccadic gaze abnormalities. In contrast, GD1 (non-neuronopathic form) tends to have more variable onset, but still typically appears in childhood, with symptoms such as hepatosplenomegaly, thrombocytopenia, anaemia and bone pain. Due to the variability in symptoms and lack of awareness, diagnosis can be significantly delayed. About 1 in 6 patients experienced a delay of ≥7 years or more from the first medical consultation to diagnosis.1
Diagnostic algorithm for pediatric patients without a family history of the disease10
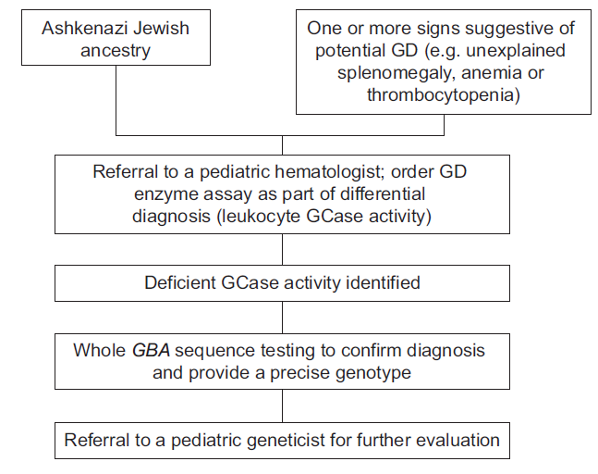
Differential diagnosis in children
Gaucher disease may be initially mistaken for:
- Hematologic malignancies1,2
- Immune thrombocytopenia4
- Thalassemia4
- Other disorders (e.g., ASMD)1,3

Would you like to know more about the diagnostics of Gaucher disease?
Would you like to know more about the diagnostics of Gaucher disease?
-
Weinreb NJ, et al. Mol Genet Metab. 2022;136(1):4-21.
-
Gupta P, et al. Expert Rev Clin Pharmacol. 2018;11(12):1183-1194.
-
Elstein D, et al. Paediatr Drugs. 2002;4(7):417-426.
-
Di Rocco M, et al. Pediatr Blood Cancer. 2014;61:1905-1909.
-
Baldellou A, et al. Eur J Pediatr. 2004;163:67-75.
-
Mistry PK, et al. Clin Adv Haematol Oncol. 2012:10:1-6.
-
Grabowski GA, et al. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Education. 2019.
-
Kaplan P, et al. Eur J Pediatr. 2013;172:447-458.
-
Gupta P, et al. Blood Cells Mol. 2011;46(1):75-84.
-
Kishnani PS, et al. Mol Genet Metab. 2021;135:154-162.
Neem contact op

MAT-BE-2600090 V1.0 03/2026