- Articolo
- Fonte: Campus Sanofi
- 4 mar 2024
Progressione e prognosi

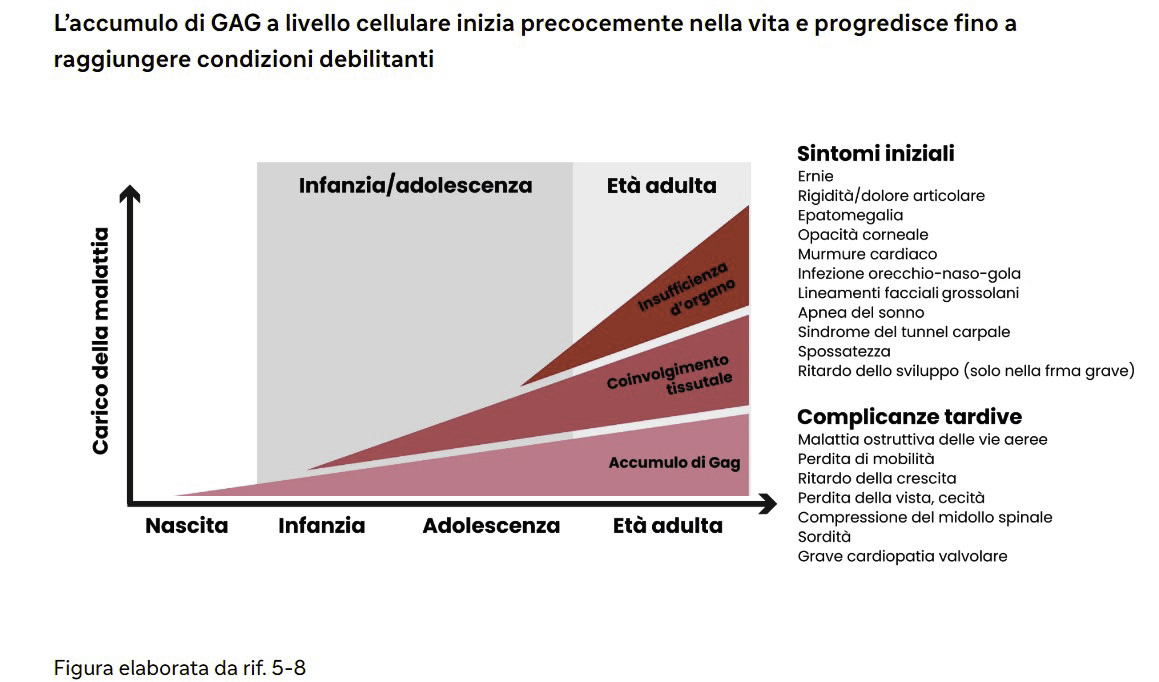
A causa dell’accumulo cronico e progressivo di glicosamminoglicani nei lisosomi cellulari in di tutto l’organismo, i pazienti affetti da MPS I manifestano una disfunzione multiorgano che causa una significativa morbilità, e una mortalità prematura nei soggetti più gravemente compromessi. Come gran parte delle altre malattie metaboliche, la MPS I è estremamente eterogenea, per cui l’età di insorgenza, i sistemi organici interessati, la gravità e il tasso di progressione della malattia variano ampiamente.1 Nei neonati e bambini affetti da malattia grave, i sintomi compaiono subito dopo la nascita e progrediscono rapidamente.1 Senza trattamento, circa il 75% di questi bambini decede prima di compiere 10 anni2 solitamente a causa di malattia ostruttiva delle vie aeree, infezioni respiratorie e complicazioni cardiache.3


Il decorso della malattia per bambini e adulti con forme attenuate di MPS I è di gran lunga più variabile. Le manifestazioni appaiono nella prima infanzia, e la progressione è più lenta e meno evidente rispetto ai soggetti affetti da malattia grave. I pazienti affetti da MPS I nella forma attenuata possono vivere fino all’età adulta, sebbene con una significativa morbilità e un’aspettativa di vita ridotta.3
L’accumulo di GAG a livello cellulare inizia precocemente nella vita e progredisce fino a raggiungere condizioni debilitanti
Leggi anche:
Bibliografia
- Beck M et al. Genet Med. 2014;16(10):759-765.
- Moore D et al. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis 2008;3:24.
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001:3421-52.
- D’Aco et al. Eur J Pediatr. 2012;171(6):911-919.
- Pastores GM et al. Mol Genet Metab 2007;91:37-47.
- Wraith JE. Expert Opin Pharmacother 2005;6:489-506.
- Vijay S, Wraith JE. Acta Pediatr 2005:94:872-7.
- Muenzer J et al. Pediatrics 2009;123:19-29.
Codice deposito aziendale MAT-IT-2100680