- Risorsa
- Fonte: Campus Sanofi
- 30 ott 2024
Un approccio multidisciplinare alla mucopolisaccaridosi I

La mucopolisaccaridosi di tipo I (MPS I) fa parte delle cosiddette malattie da accumulo lisosomiale ereditarie. Queste malattie sono caratterizzate dall’accumulo di alcune sostanze di scarto all’interno dei lisosomi.
Lisosomi: organelli cellulari che normalmente digeriscono le parti della cellula e i materiali provenienti dall’esterno diventati inutili o dannosi.
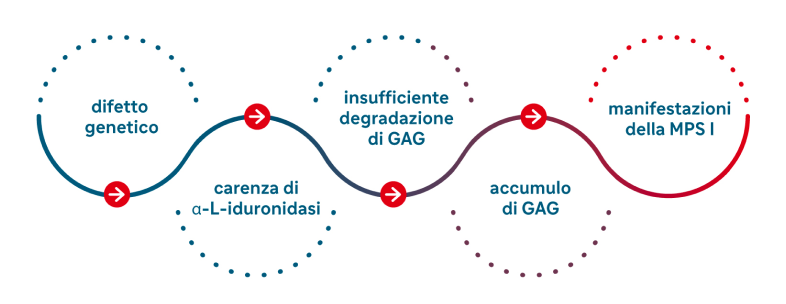
La MPS I è causata da un difetto genetico che determina la carenza dell’α-L-iduronidasi, un enzima normalmente presente nei lisosomi, che ha il compito di degradare due glicosaminoglicani (GAG): l’eparansolfato e il dermatansolfato. Quando l’α-L-iduronidasi è carente o assente, i due GAG non vengono adeguatamente eliminati e si accumulano nei lisosomi dei vari tessuti, impedendone la regolare crescita e alterandone le caratteristiche.
Sottotipi della mucopolisaccaridosi I
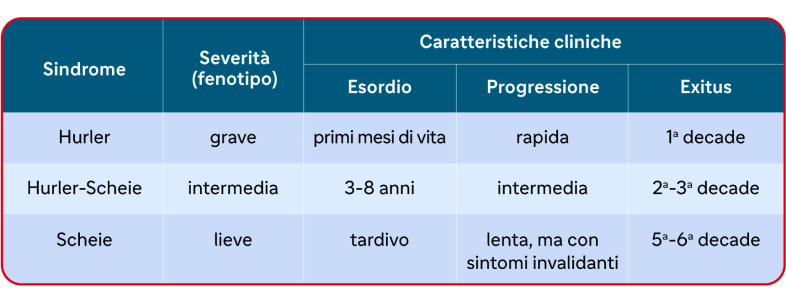
In base a queste caratteristiche, la MPS I può essere suddivisa in tre sottotipi denominati sindromi di Hurler, Hurler-Scheie e di Scheie (tabella 2) che però spesso si sovrappongono in un continuum di fenotipi clinici. Perciò ogni paziente deve essere valutato in base alle caratteristiche individuali della sua patologia e alle sue comorbidità.
Red flags per l’identificazione della MPS I
Poiché le manifestazioni cliniche della MPS I sono eterogenee e coinvolgono diversi organi, la gestione della malattia richiede un approccio multidisciplinare.
Più precoce è l’intervento dei diversi specialisti nel contribuire alla diagnosi e all’inizio tempestivo delle terapie, più elevate sono le possibilità di preservare un maggior numero di funzioni.
Principali red flags per l’identificazione della MPS I
- Infezioni precoci recidivanti dell’orecchio o delle alte vie aeree
- Epatosplenomegalia
- Lineamenti facciali grossolani e macrocefalia
- Bassa statura disarmonica
- Gibbo
- Disostosi multipla
- Ritardo nello sviluppo neurocognitivo
- Bassa statura disarmonica anche isolata, deflessione della curva di crescita
- Assente o attenuato spurt puberale
- Rigidità articolare in flessione
- Limitato range di movimento in particolare delle mani
- Dolori ricorrenti senza flogosi
- Difficoltà nei movimenti fini
- Mani ad artiglio
- Displasia dell’anca
- Cifoscoliosi
- Gibbo
- Collo corto
- Valgismo delle ginocchia
- Sindrome del tunnel carpale
- Ipotonia
- Ritardo neurocognitivo
- Regressione psicomotoria
- Opacità corneale
- Glaucoma
- Degenerazione retinica
- Mongolian spots
- Adenoidectomia, tonsillectomia (interventi multipli)
- Ernioplastica
- Rinite cronica
- Otiti ricorrenti
- Ipoacusia mista
- Ipertrofia adeno-tonsillare recidivante
- Macroglossia
- Stenosi delle vie aeree superiori
- Soffio cardiaco
- Cardiomiopatia
- Aritmia
- Valvulopatia (rigurgito aortico e mitralico, stenosi valvolare)
- Insufficienza cardiaca congestizia
- Ipertensione polmonare e sistemica
- Coronaropatia
- Cuore polmonare
Manifestazioni
La MPS I si manifesta a livello

muscolo-scheletrico
cardiologo

gastrointerinale

neurologico

uditivo
respiratorio

oculare
con diversi sintomi, gradi di severità, età di esordio, velocità di progressione, malattie concomitanti.
L’accumulo di GAG a livello di ossa, cartilagini, tendini, muscoli, cute e legamenti determina:
- patologia articolare
- artropatia, limitazione progressiva del movimento delle articolazioni fino a rigidità con dolore articolare. È tipica la “mano ad artiglio” e la sindrome del tunnel carpale,
- contratture in flessione con limitazione del movimento degli arti. È tipica la “camminata sulle punte” e la necessità di usare precocemente la sedia a rotelle,
- disostosi multipla
- rachide: gibbo lombare, cifosi dorso-lombare, scoliosi, cifoscoliosi,
- ginocchia: valgismo,
- clavicole: corte, ispessite e morfologicamente irregolari,
- anche: deformità articolari, displasia,
- ossa lunghe: accorciate e con diafisi allargate, metafisi irregolari ed epifisi non ben sviluppate,
- mani: deformità metacarpali, dita multiple a scatto,
- statura inferiore alla norma (nei pazienti Hurler a partire dai 4 anni d’età, nei quali si presenta anche collo corto).
L’accumulo di GAG nei tessuti molli oro-facciali e nelle ossa facciali determina, in modo evidente nei pazienti Hurler:
- dismorfismi facciali,
- alterazioni morfologiche del cranio come macrocefalia, microcefalia o scafocefalia.
Le manifestazioni cardiologiche sono comuni a tutte le forme di MPS I e si aggravano con l’età. Un soffio cardiaco viene talvolta riscontrato nei primi mesi di vita e riferito come primo segno della malattia.
I pazienti presentano cardiomiopatia inizialmente di tipo ipertrofico e in seguito di tipo dilatativo, con rischio di:
- aritmia
- insufficienza cardiaca congestizia,
- ipertensione polmonare e sistemica,
- cuore polmonare (insufficienza ventricolare destra).
Possono formarsi placche nell intima dell aorta e delle arterie sistemiche.
L’infiltrazione di GAG nelle arterie coronarie determina:
- ridotta funzionalità ventricolare,
- coronaropatia atipica con stenosi di vasi interi.
La frequente ipertrofia ventricolare può portare forme di scompenso cardiaco.
L’accumulo di GAG nei lembi delle valvole del cuore causa il loro progressivo ispessimento determinando:
- valvulopatia con rigurgito mitralico, meno frequentemente aortico, e stenosi valvolare.
L’accumulo di GAG nel fegato e nella milza determina epatosplenomegalia (ingrossamento del fegato e della milza) a partire dai 6 mesi.
Il fegato e la milza ingrossati comprimono l’intestino, quindi il transito gastrointestinale può essere alterato causando dolore, sazietà precoce e stitichezza alternata a diarrea.
- accumulo di GAG nelle cellule dei gangli enterici,
- anomalie della motilità intestinale,
- aumentata sensibilità alle infezioni intestinali.
Inoltre si riscontrano frequentemente voluminose ernie inguinali già a 6 mesi di età, o ernie ombelicali tra i 6 e i 12 mesi.
L’accumulo di GAG a livello di neuroni, microglia e meningi determina declino neurocognitivo che si verifica prevalentemente nella sindrome di Hurler.
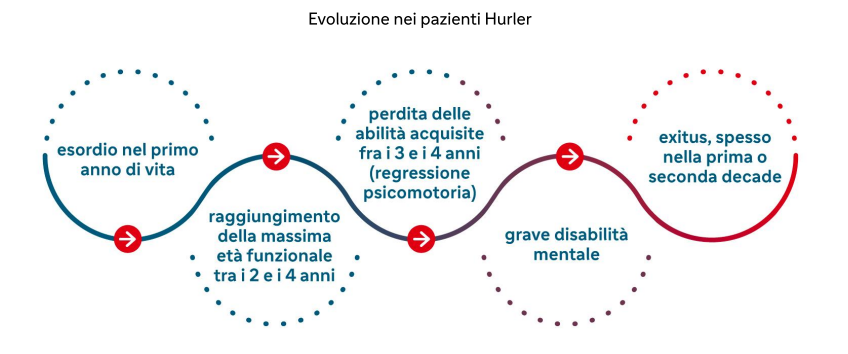
Altre manifestazioni cliniche:
- grave ritardo del linguaggio,
- ipoacusia,
- andatura anomala, alterazioni sensoriali o debolezza degli arti inferiori sino alla paralisi dovuta a mielopatia cervicale,
- limitazioni funzionali della mano e sindrome del tunnel carpale causata dalla ridotta velocità di conduzione nervosa frequenti già nell’infanzia o adolescenza. Sono frequenti anche nei pazienti Scheie adulti.
Complicanze:
idrocefalo comunicante, con conseguente aumento della pressione intracranica che può contribuire al ritardo nello sviluppo neuromotorio.
Manifestazioni cliniche:
- nausea,
- vomito,
- incontinenza,
- cecità improvvisa,
- disturbi comportamentali,
- perdita delle funzioni motorie e neurocognitive,
- cefalee ricorrenti.
L’accumulo di GAG nell’orofaringe determina l’ingrossamento delle tonsille e delle adenoidi già dal primo anno di vita con recidive all’asportazione nei pazienti Hurler. Questo contribuisce a complicanze e ostruzioni delle vie aeree superiori.
Si posssono avere anche:
- riniti croniche recidivanti accompagnate da frequenti otiti,
- ispessimento delle corde vocali,
- macroglassia,
- tracheobroncomalacia
Queste condizioni contribuiscono all’insorgenza di broncopatia ostruttiva e di complicanze a carico delle vie respiratorie come ostruzione acuta o collasso.
Lepatosplenomegalia, le deformità della colonna e le dimensioni ridotte della gabbia toracica con stenosi tracheali multiple e ridotta capacità polmonare determinano:
- respirazione notturna rumorosa,
- apnee ostruttive,
- sindrome ipoventilatoria,
- dispnea,
- atelectasia polmonare,
- ipertensione polmonare.
Nei pazienti Scheie, le principali affezioni a carico delle vie respiratorie superiori consistono in rinosinusiti croniche recidivanti e frequenti otiti medie.
Nei pazienti Scheie, la ridotta capacità polmonare è un parametro da monitorare nel tempo.
Le complicanze respiratorie rappresentano una delle principali cause di morte nei pazienti con MPS I.
Una manifestazione oculare frequente è l’opacità corneale diffusa, talvolta associata a fotosensibilità e ridotta acuità visiva.
L’accumulo di GAG nel tessuto trabecolare e nella sclera porta all’aumento della pressione intraoculare.
Nei pazienti Hurler, il glaucoma, oltre all’aumento della pressione intracranica e all’idrocefalo comunicante, si può sviluppare atrofia del nervo ottico. Inoltre, si può verificare degenerazione retinica.
Anche i pazienti Scheie possono presentare alterazioni oculari sino al glaucoma.
L’accumulo di GAG negli ossicini dell’orecchio medio e nel nervo acustico determina ipoacusia.
I pazienti Hurler presentano ipoacusia di natura
- conduttiva o
- neurosensoriale o
- mista
anche a causa delle frequenti infezioni e lesioni della membrana timpanica. Otiti medie possono comparire già a 6 mesi di età.
Anche i pazienti Scheie presentano ipoacusia, più comunemente alle alte frequenze, e ricorrenti otiti.
L’ipoacusia può implicare significative difficoltà nelle interazioni sociali, nello sviluppo del linguaggio, cognitivo e comportamentale.
Algoritmo diagnostico per la forma di MPS I attenuata nei bambini sopra i 5 anni
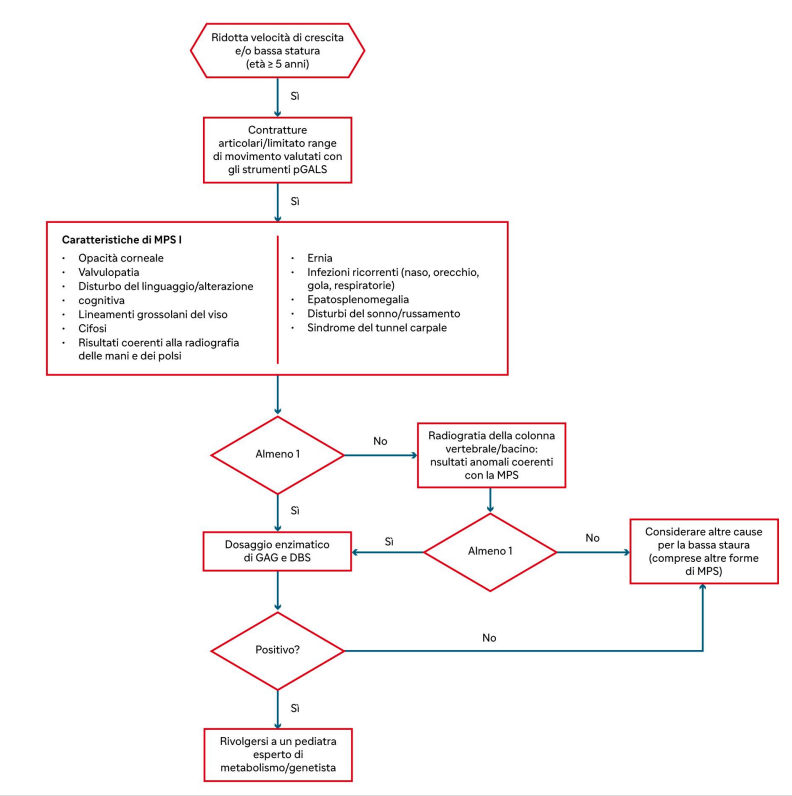
Diagnosi
La diagnosi precoce è fondamentale per permettere al paziente di iniziare al più presto la terapia preservando il maggior numero di funzioni.
Un ritardo diagnostico riguarda soprattutto i fenotipi meno gravi, caratterizzati da sintomi poco specifici.
I pazienti che presentano contratture spesso ricevono la diagnosi di:
- artrite reumatoide,
- artrite idiopatica giovanile,
- sclerodermia.
Il sospetto di MPS I dovrebbe sorgere quando:
- le contratture non sono associate a gonfiore o infiammazione locale,
- le contratture non sono inizialmente dolenti,
- la sintomatologia non risponde al trattamento anti-infiammatorio,
- non si rileva evidenza radiologica di segni di erosione.

DBS = spot di sangue secco; GAG = glicosaminoglicani; MPS I = mucopolisaccaridosi di tipo I; pGALS = paediatric Gait, Arms, Legs and Spine; u-GAG = GAG urinari
Bibliografia
Gasperini S, Scarpa M, Spada M. Un approccio multidisciplinare alla mucopolisaccaridosi I: dall’identificazione precoce alla terapia avanzata. AboutMedicine Anno XII, N. 7, giugno 2024
Codice deposito aziendale MAT-IT-2402618