- Articolo
- Fonte: Campus Sanofi
- 11 mar 2024
Presentazione clinica

La MPS I è storicamente suddivisa in tre fenotipi definiti in modo soggettivo che comprendono il fenotipo grave a rapida progressione (sindrome di Hurler) e i fenotipi attenuati a progressione più lenta (sindromi di Hurler-Scheie e Scheie).1
_0.2023-12-28-08-43-06.png)
Caratteristiche delle forme di MPS I2-4

Tuttavia, è presente una sostanziale sovrapposizione della sintomatologia tra i fenotipi e ogni paziente affetto presenta un decorso clinico distinto e una serie di sintomi univoca.5
Di seguito potrai avere una panoramica completa circa i fenotipi e i sintomi caratteristici delle forme Hurler, Hurler-Scheie e Scheie:
MPS I forma grave: Hurler

Aleksandra | MPS I grave (Hurler) |Polonia
Aleksandra | MPS I grave (Hurler) |Polonia
I bambini affetti da MPS I in forma grave (Hurler) presentano evidenti anomalie fisiche e cognitive subito dopo la nascita che peggiorano rapidamente, tanto che la maggior parte dei pazienti non trattati decede entro la prima decade di vita. Presentano un notevole ritardo dello sviluppo e una compromissione cognitiva, oltre a caratteristici lineamenti facciali, deformità scheletriche, e problemi di carattere respiratorio, cardiaco ed epatico.6
Segni più comuni*6,7
- Ernie ricorrenti (ombelicali/inguinali).
- Cifosi/deformazione gibbosa.
- Lineamenti facciali grossolani.
- Opacità corneale.
- Epatosplenomegalia.
- Disfunzione cognitiva.
- Disturbi del sonno/russamento.
- Anomalie della valvola cardiaca.
- Bassa statura.
*Non tutti i segni e sintomi vengono osservati in tutti i pazienti. [elenco estrapolato da rif. 6, figura 2, sintomi che si manifestano in più del 50% dei pazienti].
Prevalenza ed età di insorgenza di segni e sintomi nei pazienti con MPS I, forma Hurler
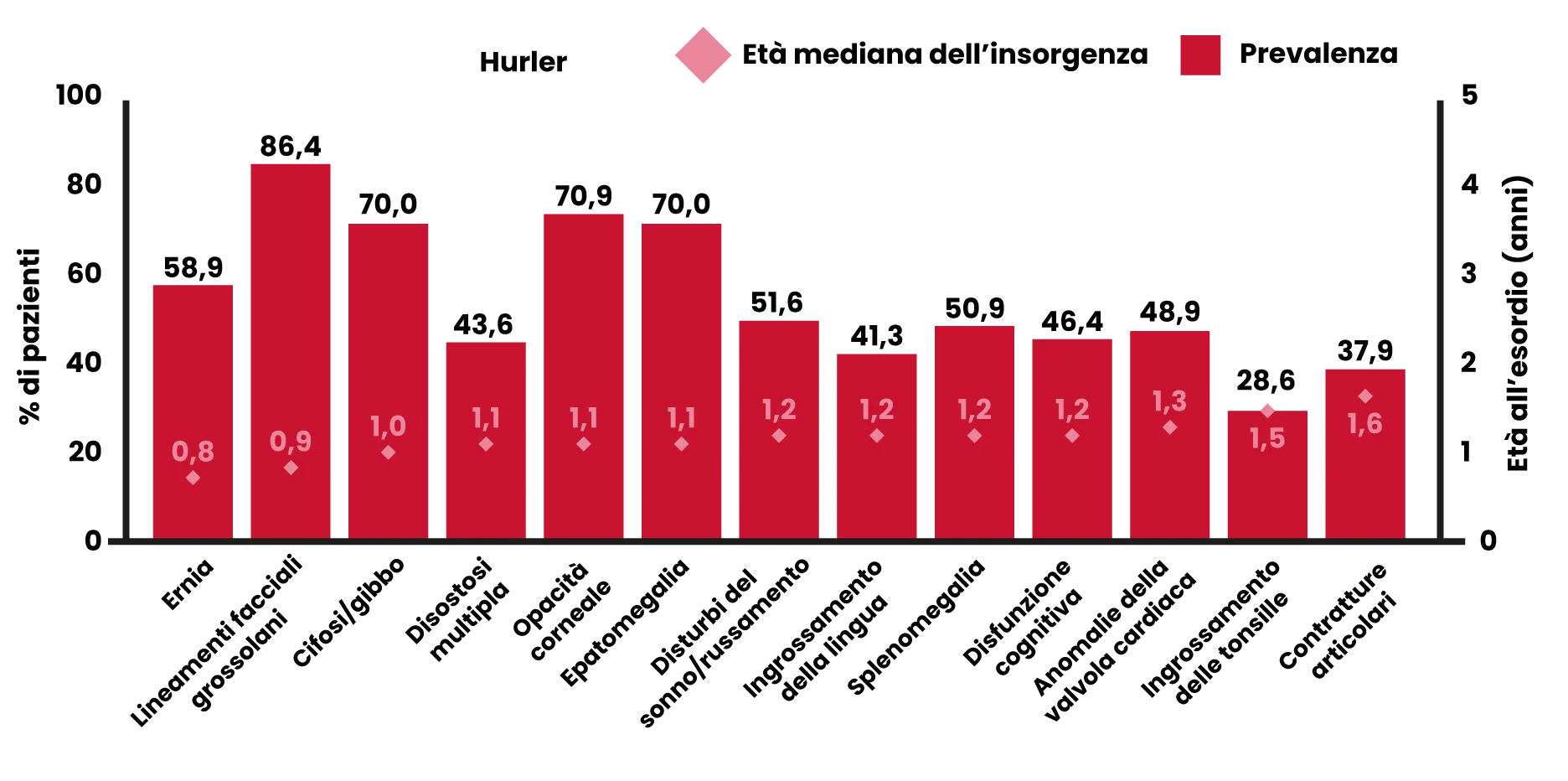
Elaborato da Fig. 2, rif 6.
MPS I forme attenuate: Hurler-Scheie e Scheie

Dounia | MPS I attenuata (Hurler-Scheie) |Francia

Tamara | MPS I attenuata | NL
Dounia | MPS I attenuata (Hurler-Scheie) |Francia
Tamara | MPS I attenuata | NL
Bambini e adulti con forme attenuate di MPS I (Hurler-Scheie e Scheie) presentano anomalie fisiche meno evidenti e disfunzione cognitiva lieve o assente. Le manifestazioni progrediscono più lentamente rispetto ai pazienti con malattia grave. Sintomi come ernie, contratture articolari e/o infezioni respiratorie in genere insorgono entro la prima decade di vita, ma possono essere uguali ai sintomi riscontrati in condizioni più comuni e ciò determina anni di ritardo diagnostico.8-10 I pazienti che si presentano con contratture articolari in assenza di infiammazione devono essere sottoposti al test per la MPS I senza ritardo. 1,9,10
Segni più comuni*6,7
- Ernie ricorrenti (ombelicali/inguinali).
- Lineamenti facciali grossolani.
- Contratture articolari.
- Opacità corneale.
- Disturbi del sonno/russamento.
- Epatomegalia.
- Anomalie della valvola cardiaca.
- Sindrome del tunnel carpale.
- Bassa statura.
*Non tutti i segni e sintomi vengono osservati in tutti i pazienti. [elenco estrapolato da rif. 6, figura 2, sintomi che si manifestano in più del 50% dei pazienti con le sindromi di Hurler-Scheie/Scheie combinate].
Prevalenza ed età di insorgenza di segni e sintomi nei pazienti con MPS I, forma Hurler-Scheie e Scheie
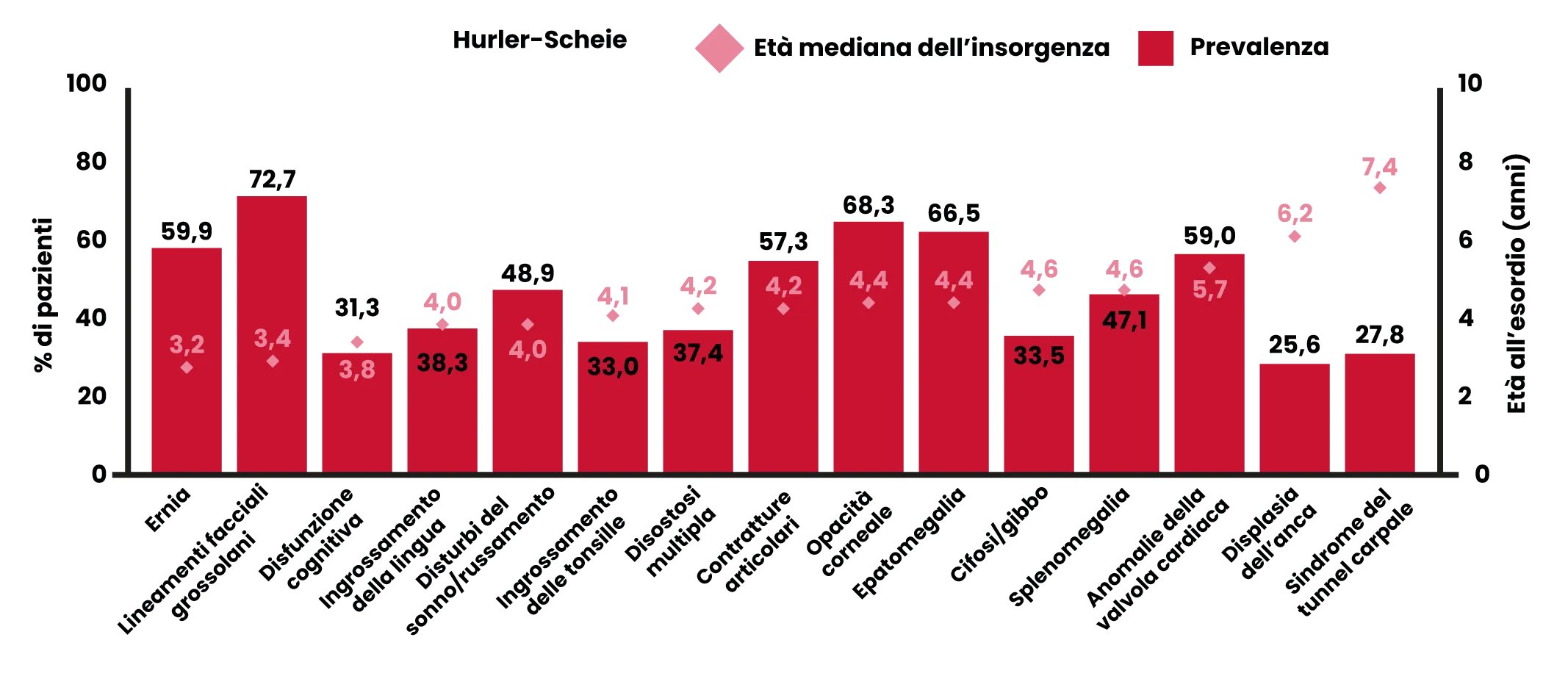
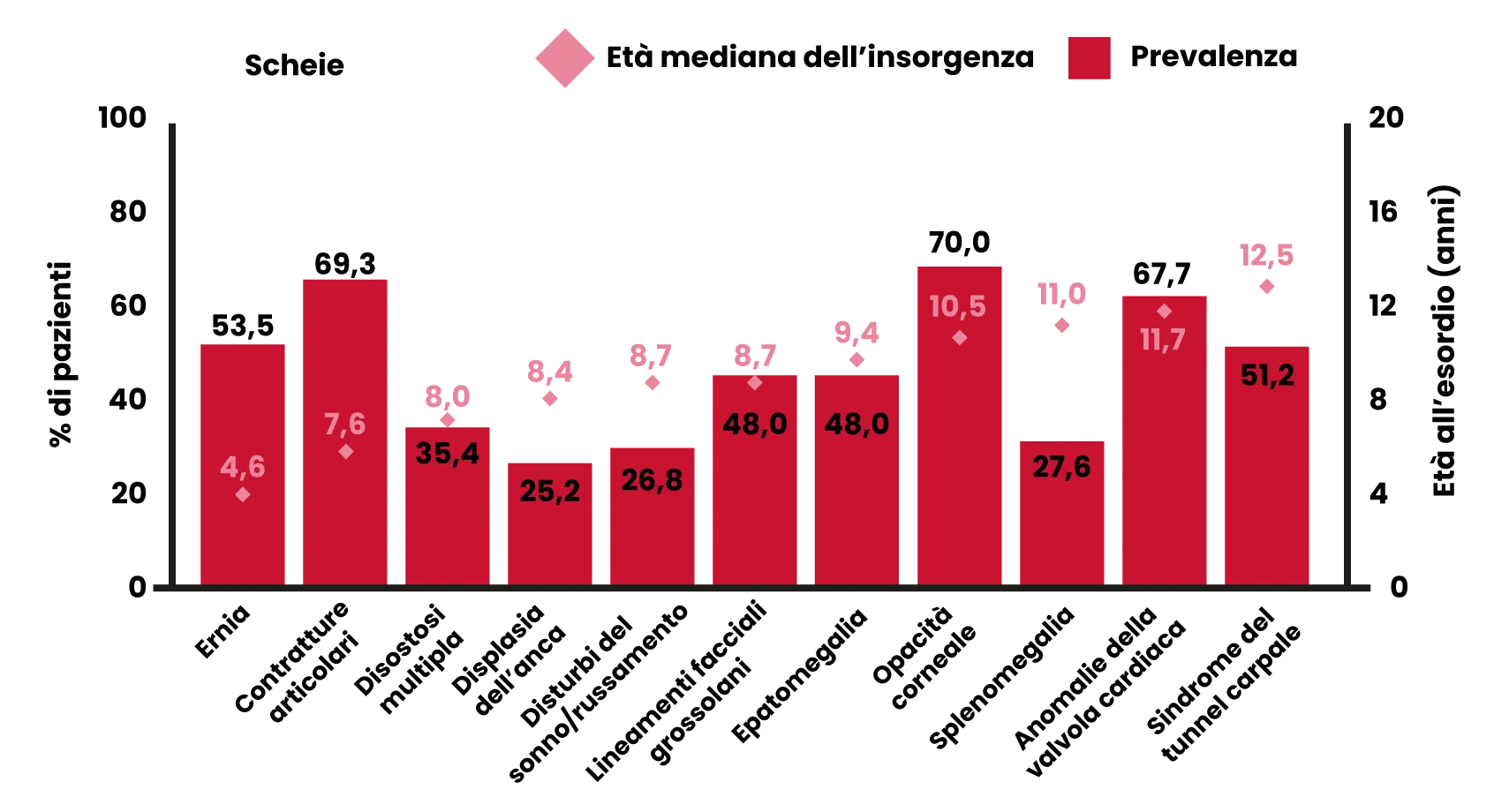
Elaborato da Fig. 2, rif 6.
Leggi anche:
Bibliografia
- Neufeld EF, Muenzer J. (2001) The mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, and Vogelstein B. (eds.). 8th edition, Vol. III. McGraw-Hill, Medical Publishing Division, pp. 3421.
- Muenzer J et al. Pediatrics 2009;123:19-29.
- Fallet S et al. JIEMS. 2014;2:5.
- Moore D et al. Orphanet J Rare Dis. 2008;3:24.
- MPS I Registry Boards of Advisors. Cambridge, MA: Genzyme Corporation; 2010.
- Beck M et al. Genet Med. 2014;16(10):759-765.
- De Ru et al. Orphanet Journal of Rare Diseases 2012, 7:22.
- Vijay S, Wraith JE. Acta Paediatr 2005;94:872–77.
- Cimaz R et al. Clin Exp Rheumatol 2006;24:196-202.
- Thomas JA et al. J Inherit Metab Dis 2010;33:421-7.
Codice deposito aziendale MAT-IT-2100680