- Article
- Source : Campus Sanofi
- 8 août 2025
Qu'est-ce que la maladie de Fabry ?

La maladie de Fabry est une maladie du métabolisme, rare et héréditaire de transmission liée au chromosome X. Elle affecte aussi bien les hommes que les femmes, mais ne se transmet pas de père à fils.
La maladie de Fabry est due à des variants pathogènes du gène GLA responsables d’un déficit d’une enzyme lysosomale : l’alpha-galactosidase A. Ce déficit enzymatique entraîne une accumulation de glycosphingolipides (Gb3 et Lyso-Gb3) dans toutes les cellules de l’organisme résultant en une affection multisystémique progressive et sévère responsable d’une morbi-mortalité élevée.
L’incidence de la maladie est estimée à 1 cas pour 10 000 naissances. Son expression est très hétérogène. Les patients peuvent présenter plusieurs atteintes (cardiaques, rénales, cutanées, du système nerveux central, du système nerveux périphérique…) ou une seule d’entre elles1.
Les signes cliniques de la maladie de Fabry peuvent être multi-systémiques et hétérogènes. Il est possible de distinguer deux phénotypes principaux2 :
• Une forme classique, où l’activité enzymatique de l’α-Gal A est très réduite ou même inexistante. Dans ce cas, les patients manifestent souvent des symptômes depuis l’enfance, qui peuvent se compliquer sur le long-terme3.
MALADIE MULTI-SYSTEMIQUE AVEC UNE ATTEINTE VISCERALE (cœur, reins, cerveau)1
• Une forme tardive, dans laquelle il existe une activité résiduelle de l’α-Gal A. Les patients atteints de cette forme peuvent être asymptomatiques ou bien développer des symptômes plus tard dans la vie, cependant, la maladie peut évoluer jusqu’à un stade sévère3.
TABLEAU CLINIQUE INCOMPLET (essentiellement cardiaque)1
Une représentation humanoïde est proposée ci-dessous afin d’illustrer l’ensemble des atteintes associées à la maladie1 :
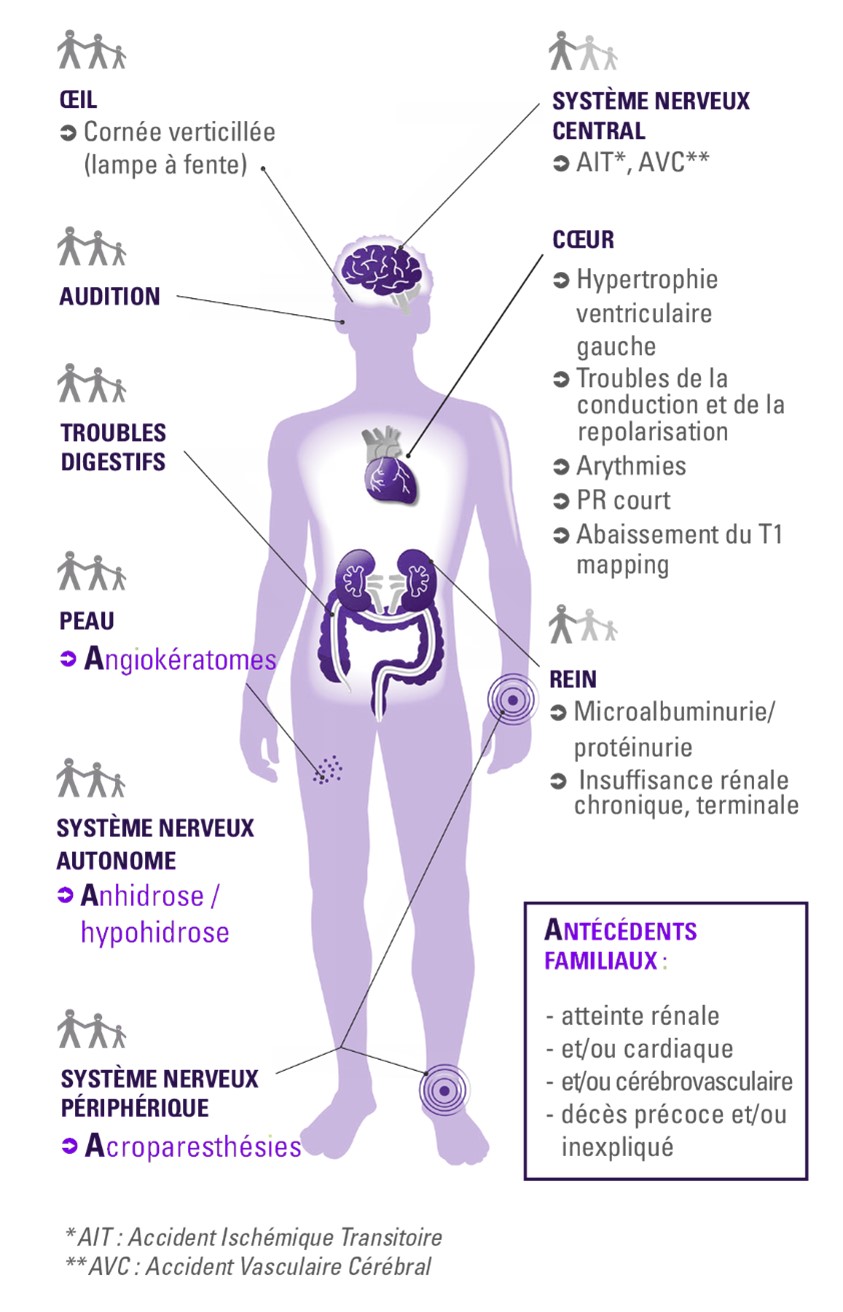
Chez l’homme, le diagnostic repose sur la mesure de l’activité enzymatique de l’α-galactosidase A dans les leucocytes (ou le papier buvard). Le séquençage du gène GLA est indispensable à la confirmation biologique du diagnostic et peut aider à la caractérisation de la maladie1.
Chez la femme, le diagnostic repose sur le séquençage du gène GLA (activité de l’αgalactosidase A pouvant être normale). Un dosage initial du lyso-Gb3 plasmatique est recommandé pour une meilleure caractérisation de la maladie, une aide à l’interprétation de la pathogénicité du variant et le suivi thérapeutique1.
En pratique : Le dosage sanguin de l'activité de l'α-galactosidase A et le dosage du lyso-Gb3 sont réalisables par l'intermédiaire de votre centre hospitalier ou bien via un laboratoire de ville*. Pour le génotypage du gène GLA, il faut joindre le formulaire de consentement éclairé du patient nécessaire à toute recherche génétique et obtenir un avis spécialisé en génétique pour interpréter la pathogénicité du variant.
Le diagnostic précoce permet un suivi optimal des patients et une prise en charge adaptée avant l’apparition de lésions sévères irréversibles.
* Examens remboursés à 100%
La prise en charge de la maladie de Fabry est complexe et nécessite une approche personnalisée pour chaque patient et une évaluation multidisciplinaire. Elle est réalisée par une équipe pluridisciplinaire d’un centre de référence / compétence de la maladie de Fabry.
On distingue la prise en charge thérapeutique et la prise en charge paramédicale. Actuellement la prise en charge thérapeutique repose sur plusieurs alternatives : la thérapie enzymatique substitutive (TES) ou une molécule chaperon. Concernant la prise en charge paramédicale, une approche diététique peut être offerte aux patients, notamment lors des symptômes digestifs gênants. Les complications chroniques d’organe (insuffisance rénale, insuffisance cardiaque) nécessitent des conseils diététiques spécifiques.
Il est également essentiel de pouvoir proposer aux familles un soutien psychologique, compte tenu de l’impact du diagnostic génétique, de l’implication des descendants et des sentiments de culpabilité, angoisse et tristesse qu’éprouvent souvent les patients à l’égard de leur maladie. L’intervention d’un psychologue peut s’avérer précieuse, notamment lors de l’annonce du diagnostic qui représente une étape souvent éprouvante pour les patients1.
S’agissant d’une maladie génétique héréditaire, l’enquête familiale est précise pour diagnostiquer précocement les membres de la famille potentiellement atteint à partir d’un patient1.
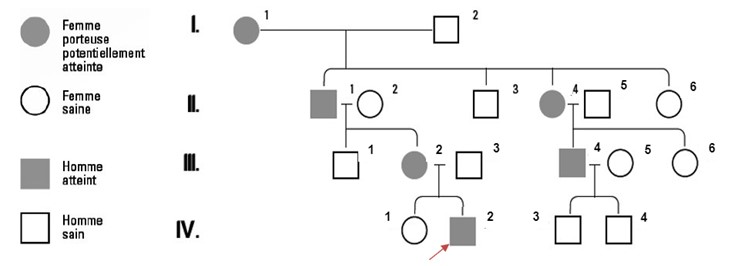
Arbre généalogique d’une famille atteinte de la maladie de Fabry
À partir d’un cas index, on retrouve en moyenne 5 autres membres de la famille atteints de maladie de Fabry (père, mère, frère, sœur, tante, oncle, cousin, cousine…), en dressant l’arbre généalogique de manière exhaustive5. Ainsi, les conseillers en génétique jouent un rôle clé dans la prise en charge du patient1.
Contactez votre conseiller en génétique via le répertoire de l’association française des conseillers en génétique : https://af-cg.fr/
Les signes cliniques de la maladie de Fabry peuvent être multi-systémiques et hétérogènes. Il est possible de distinguer deux phénotypes principaux2 :
• Une forme classique, où l’activité enzymatique de l’α-Gal A est très réduite ou même inexistante. Dans ce cas, les patients manifestent souvent des symptômes depuis l’enfance, qui peuvent se compliquer sur le long-terme3.
MALADIE MULTI-SYSTEMIQUE AVEC UNE ATTEINTE VISCERALE (cœur, reins, cerveau)1
• Une forme tardive, dans laquelle il existe une activité résiduelle de l’α-Gal A. Les patients atteints de cette forme peuvent être asymptomatiques ou bien développer des symptômes plus tard dans la vie, cependant, la maladie peut évoluer jusqu’à un stade sévère3.
TABLEAU CLINIQUE INCOMPLET (essentiellement cardiaque)1
Une représentation humanoïde est proposée ci-dessous afin d’illustrer l’ensemble des atteintes associées à la maladie1 :
Chez l’homme, le diagnostic repose sur la mesure de l’activité enzymatique de l’α-galactosidase A dans les leucocytes (ou le papier buvard). Le séquençage du gène GLA est indispensable à la confirmation biologique du diagnostic et peut aider à la caractérisation de la maladie1.
Chez la femme, le diagnostic repose sur le séquençage du gène GLA (activité de l’αgalactosidase A pouvant être normale). Un dosage initial du lyso-Gb3 plasmatique est recommandé pour une meilleure caractérisation de la maladie, une aide à l’interprétation de la pathogénicité du variant et le suivi thérapeutique1.
En pratique : Le dosage sanguin de l'activité de l'α-galactosidase A et le dosage du lyso-Gb3 sont réalisables par l'intermédiaire de votre centre hospitalier ou bien via un laboratoire de ville*. Pour le génotypage du gène GLA, il faut joindre le formulaire de consentement éclairé du patient nécessaire à toute recherche génétique et obtenir un avis spécialisé en génétique pour interpréter la pathogénicité du variant.
Le diagnostic précoce permet un suivi optimal des patients et une prise en charge adaptée avant l’apparition de lésions sévères irréversibles.
* Examens remboursés à 100%
La prise en charge de la maladie de Fabry est complexe et nécessite une approche personnalisée pour chaque patient et une évaluation multidisciplinaire. Elle est réalisée par une équipe pluridisciplinaire d’un centre de référence / compétence de la maladie de Fabry.
On distingue la prise en charge thérapeutique et la prise en charge paramédicale. Actuellement la prise en charge thérapeutique repose sur plusieurs alternatives : la thérapie enzymatique substitutive (TES) ou une molécule chaperon. Concernant la prise en charge paramédicale, une approche diététique peut être offerte aux patients, notamment lors des symptômes digestifs gênants. Les complications chroniques d’organe (insuffisance rénale, insuffisance cardiaque) nécessitent des conseils diététiques spécifiques.
Il est également essentiel de pouvoir proposer aux familles un soutien psychologique, compte tenu de l’impact du diagnostic génétique, de l’implication des descendants et des sentiments de culpabilité, angoisse et tristesse qu’éprouvent souvent les patients à l’égard de leur maladie. L’intervention d’un psychologue peut s’avérer précieuse, notamment lors de l’annonce du diagnostic qui représente une étape souvent éprouvante pour les patients1.
S’agissant d’une maladie génétique héréditaire, l’enquête familiale est précise pour diagnostiquer précocement les membres de la famille potentiellement atteint à partir d’un patient1.
Arbre généalogique d’une famille atteinte de la maladie de Fabry
À partir d’un cas index, on retrouve en moyenne 5 autres membres de la famille atteints de maladie de Fabry (père, mère, frère, sœur, tante, oncle, cousin, cousine…), en dressant l’arbre généalogique de manière exhaustive5. Ainsi, les conseillers en génétique jouent un rôle clé dans la prise en charge du patient1.
Contactez votre conseiller en génétique via le répertoire de l’association française des conseillers en génétique : https://af-cg.fr/
Références
- Protocole National de Diagnostic et de Soins Maladie de Fabry. HAS. Novembre 2021.
- Protocole national de diagnostic et de soins Maladie de Fabry. HAS. Novembre 2021.
- Miller J.J. et al. Progress in the understanding and treatment of Fabry disease. Biochim Biophys Acta - General Subjects. 2020; 1864(1), 129437.
- Germain D.P. et al. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol Genet Metab. 2022; 137(1-2),49-61
- Eng. C.M. et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J. Inherit. Metab. Dis. 2007;30:184-92.
- Germain et al. The benefits and challenges of family genetic testing in rare genetic diseases— l essons from Fabry disease 2021; 9:e1666
250709143503XD – 09/2025