- Articolo
- Fonte: Campus Sanofi
- 15 mag 2024
NPD: Definizione e Classificazione

La NPD è storicamente classificata in 3 sottotipi: NPD A, NPD B, e NPD C.1
- NPD A e B sono conosciute anche come malattie da deficit di sfingomielinasi acida (o ASMD) e sono causate da mutazioni del gene sfingomielina fosfodiesterasi 1 (SMPD1).2,3
- NPD C è un disordine di esterificazione del colesterolo, causato da mutazioni dei geni NPC1 o NPC2.2,4
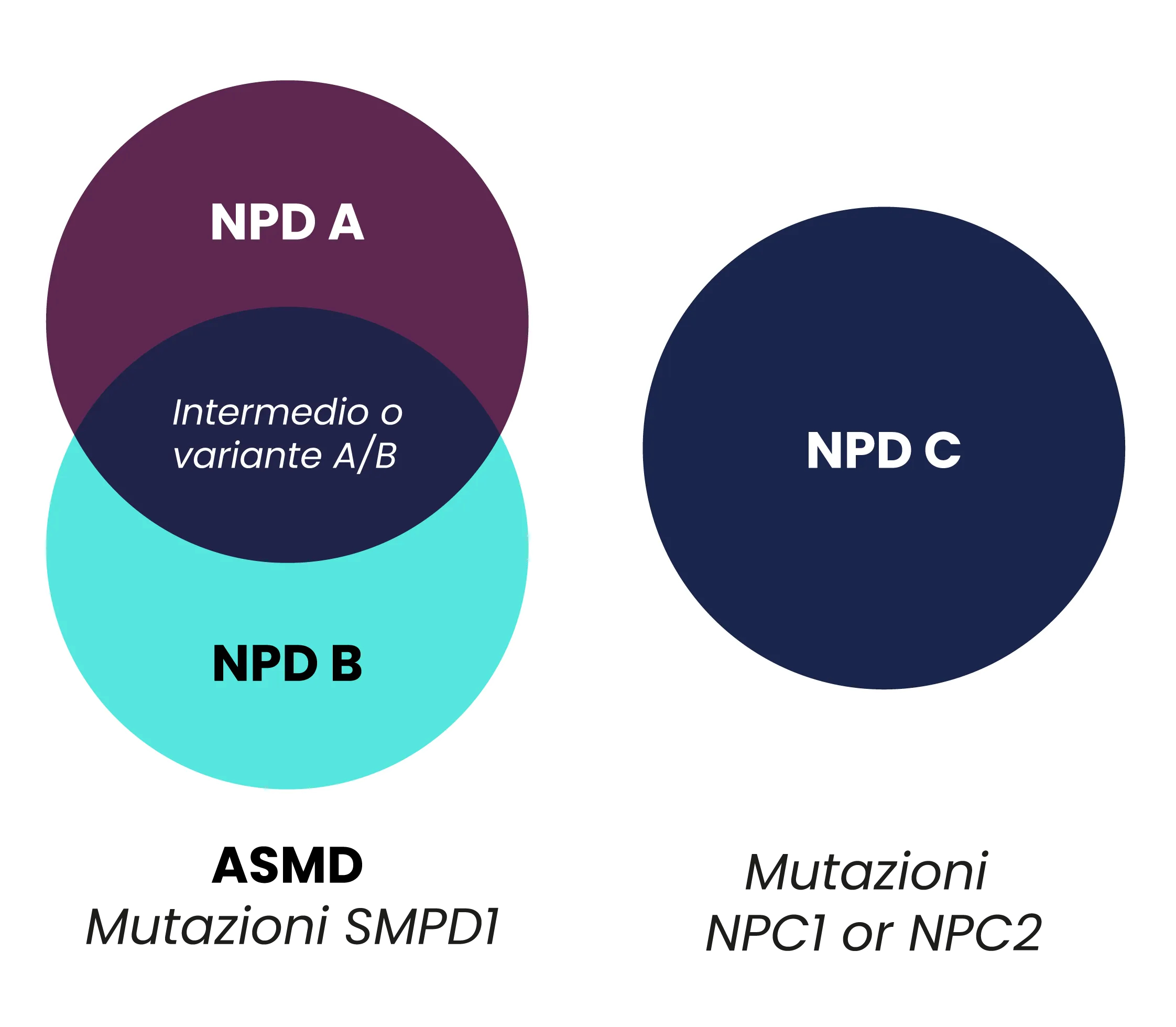
Storicamente l’ASMD era suddivisa in 2 sottocategorie della malattia di Niemann-Pick in base alla presenza (NPD tipo A) o assenza (NPD tipo B) di un coinvolgimento neurologico.5
Successivamente è stata aggiunta una terza categoria con fenotipo intermedio.6
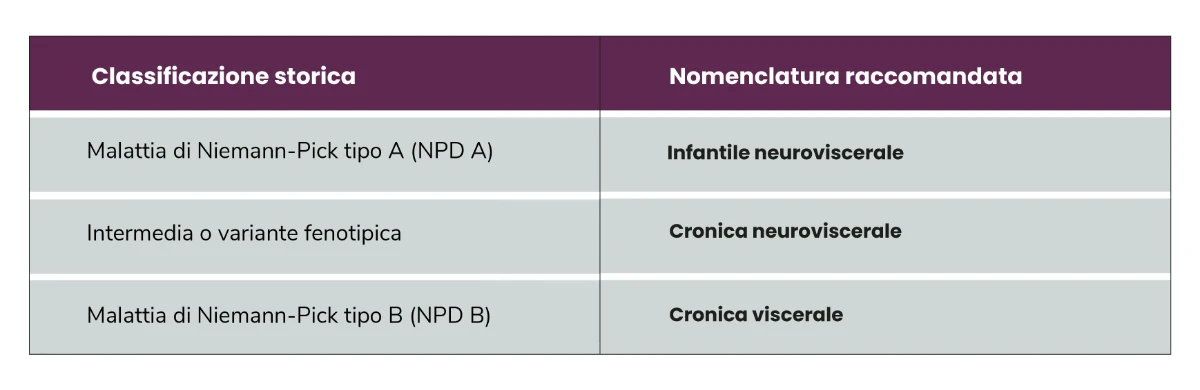
Oggi si raccomanda un diverso tipo di classificazione:
Il deficit di sfingomielinasi acida (ASMD) è una malattia autosomica recessiva, grave e progressiva. Storicamente nota anche come malattia di Niemann-Pick (NP) di tipo A, A/B e B, l'ASMD è un disordine da accumulo lisosomiale progressivo e multisistemico che può colpire pazienti pediatrici e adulti.7
L'ASMD è una malattia da accumulo lisosomiale causata da varianti patogene del gene sfingomielina fosfodiesterasi 1 (SMPD1) codificante per l'enzima ASM.7
L'insorgenza e la progressione della malattia sono eterogenei e possono portare a complicanze multiorgano.7
Segni e sintomi si manifestano comunemente a carico della milza, del fegato, dei polmoni e del sistema ematologico; in alcune tipologie di pazienti viene coinvolto anche il sistema nervoso.7
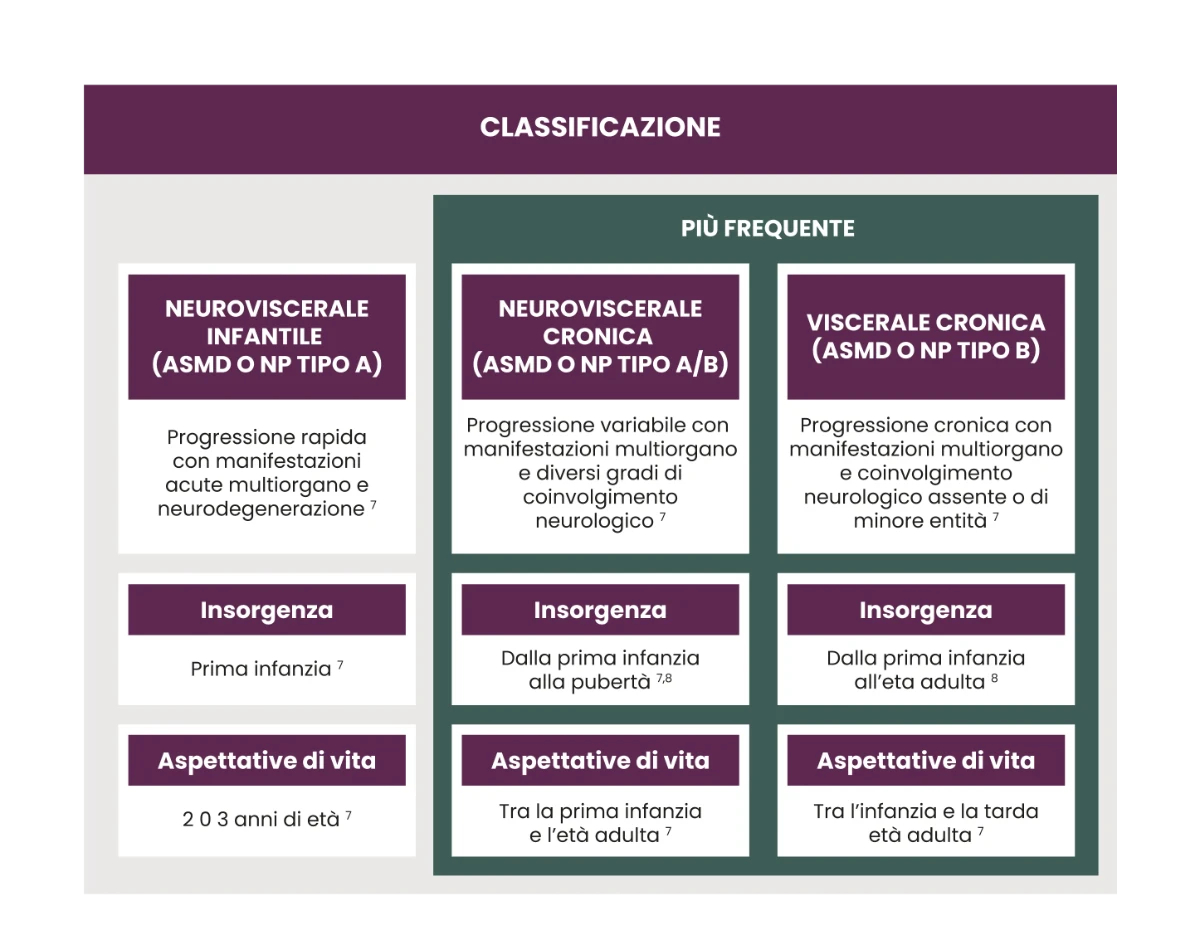
L’ASMD presenta un ampio spettro clinico di malattia, diviso in 3 tipi:7
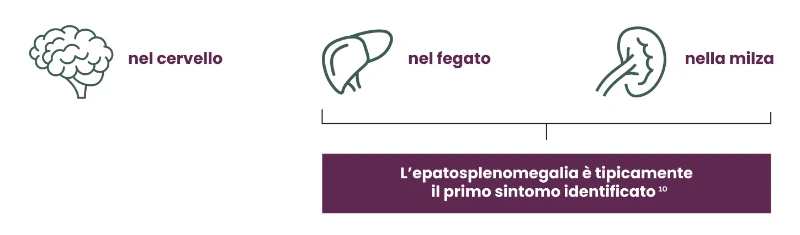
La sfingomielina si accumula principalmente:9,10

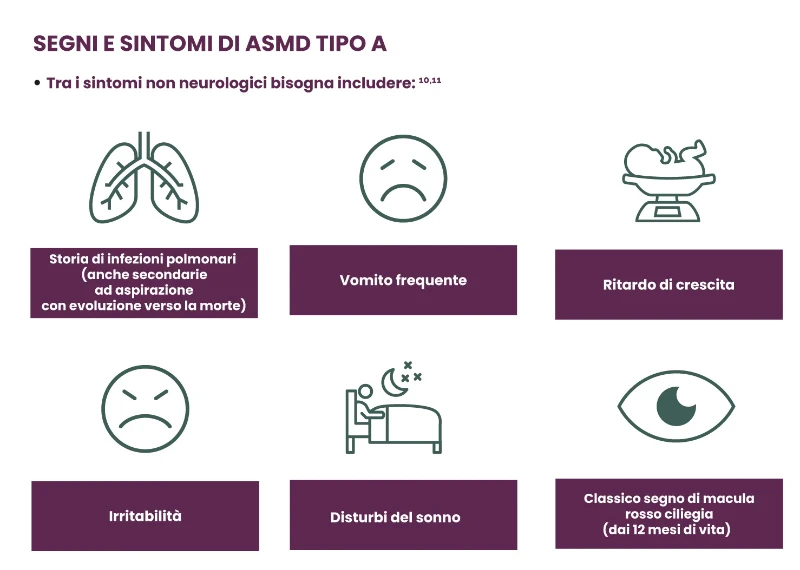
- Il fenotipo A/B è caratterizzato da manifestazioni sistemiche simili o più severe rispetto al fenotipo B associate ad una lenta e progressiva neurodegenerazione.12
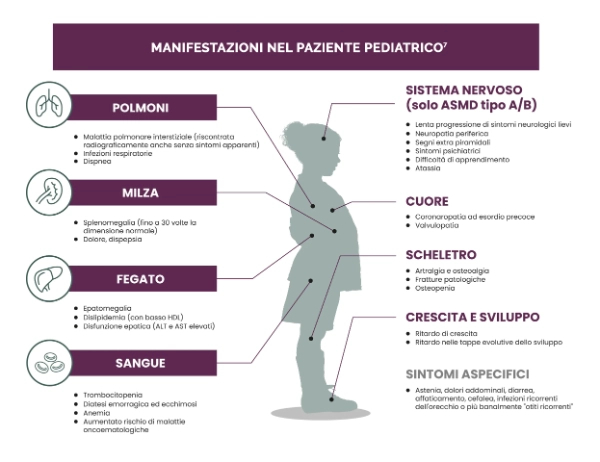
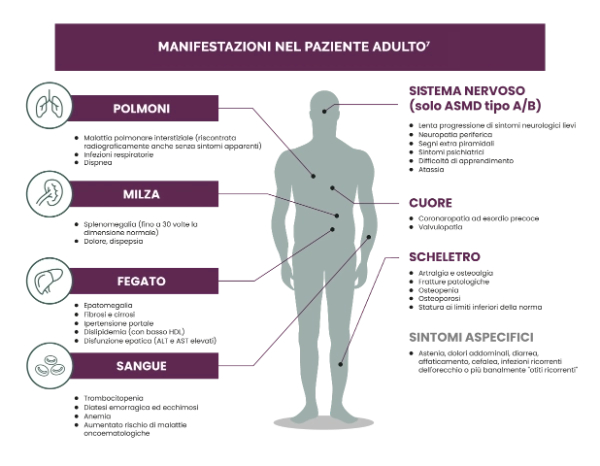
Le manifestazioni di ASMD possono limitare la capacità dei pazienti di operare, compromettendo la loro vita personale e le loro prospettive per il futuro.7,13
- Frequente necessità di ricovero, assunzione constante di farmaci per trattare i sintomi, ricorso a procedure mediche e dispositivi medici.
- Attività fisica limitata e incapacità di soddisfare le attività scolastiche, lavorative e relative alle relazioni personali.
- Sensazioni di isolamento sociale e di rifiuto.
Indipendentemente dallo spettro di malattia che i pazienti presentano, l’ASMD può avere un impatto negativo sul benessere e sulla qualità della loro vita: è importante riconoscere i principali segni e sintomi e sospettare l'ASMD tempestivamente.
Per prevenire il ritardo diagnostico LEGGI L’APPROFONDIMENTO
Leggi anche:
Bibliografia
- Vanier MT. Chapter 176 - Niemann–Pick diseases. In: Olivier Dulac ML, Harvey BS, editors. Handbook of Clinical Neurology. Volume 113: Elsevier; 2013. p. 1717-21.
- Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: Acid sphingomyelinase deficiencies. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). New York, NY: McGraw-Hill. Chapt 144.
- Desnick JP et al. Mol Med. 2010;16:316-321.
- McGovern MM et al. Pediatrics. 2008;122(3):e341-e349.
- Pentchev PG et al. Proc Natl Acad Sci U S A. 1985 Dec;82(23):8247-51.
- Wasserstein MP et al. J Pediatr. 2006 Oct;149(4):554-9.
- McGovern MM et al. Orphanet J Rare Dis. 2017;12:41.
- Cassiman D et al. Mol Genet Metab. 2016;118(3):206-213.
- Ledesma MD et al. J Neurochem. 2011 Mar;116(5):779-88.
- McGovern MM et al. Neurology. 2006;66(2):228-232.
- Schuchman EH. Inherit Metab Dis. 2007 Oct;30(5):654-63.
- McGovern MM et al. Genet Med. 2017;19(9)967-974.
- Henderson SL et al. Am J Med Genet A. 2009;149A(11):2430-2436
Codice deposito aziendale MAT-IT-2302491