- Articolo
- Fonte: Campus Sanofi
- 8 apr 2024
Patogenesi della MPS I

È una malattia ereditaria, autosomica recessiva con manifestazioni patologiche nella maggior parte dei sistemi organici e dei tessuti. La malattia è causata da mutazioni del gene che codifica l’enzima lisosomiale α-L-iduronidasi, per cui i soggetti affetti non sono in grado di produrre l’enzima o lo producono in basse quantità. Ciò compromette la capacità delle cellule di degradare i glicosamminoglicani (GAG) dermatan ed eparan solfato, determinando un accumulo progressivo di GAG nelle cellule.1 Questo scatena una cascata di processi patologici organo-specifici, alterazioni cellulari e tissutali, che solitamente diventano irreversibili con il progredire della malattia.3
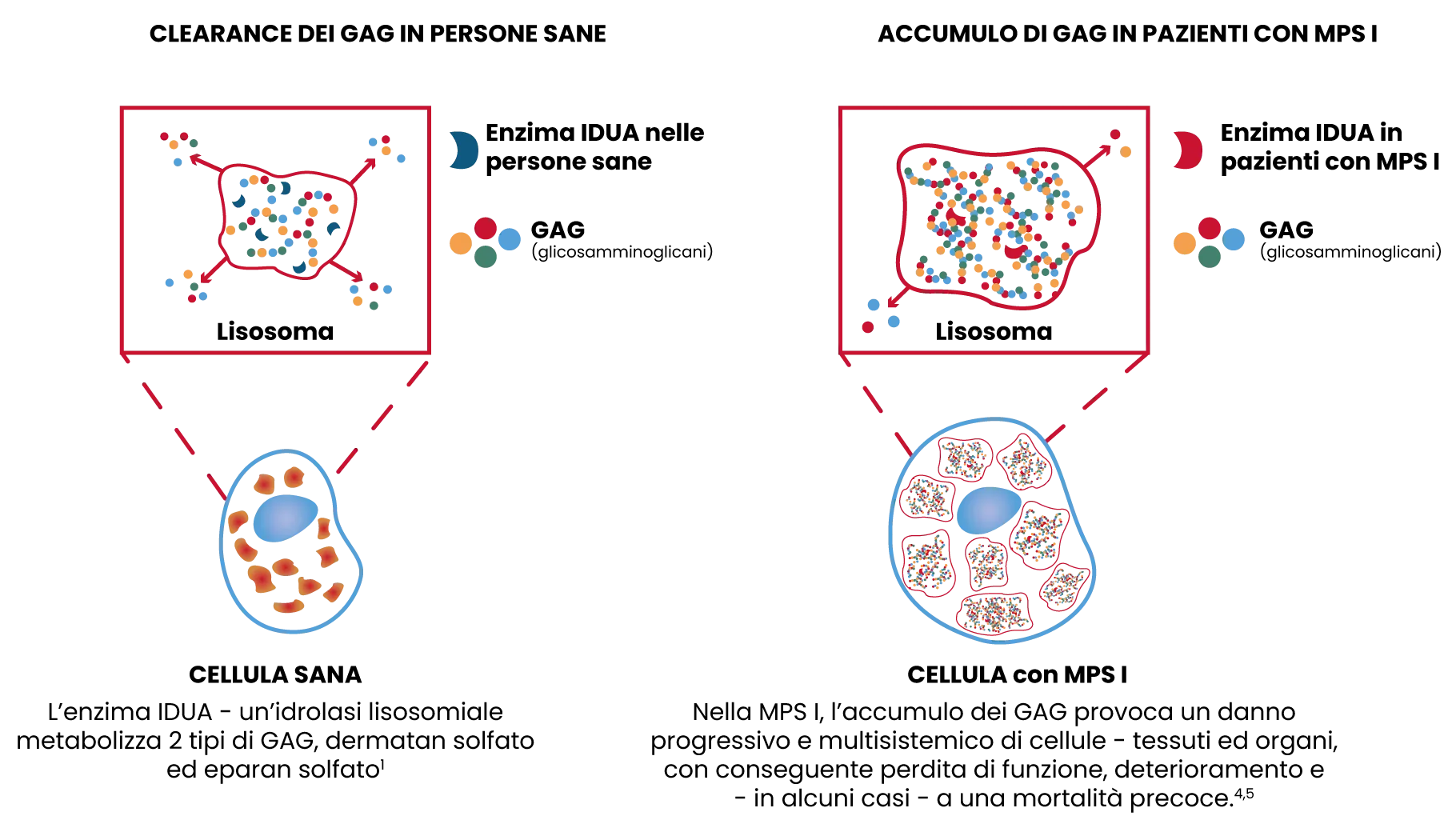
Alla nascita, i bambini affetti dalla malattia inizialmente sembrano essere clinicamente normali, ma con il passare del tempo, il progressivo accumulo di GAG causa un peggioramento delle funzionalità motorie, respiratorie e cardiache, ed è responsabile dell’ingrossamento di organi come fegato e milza.1,6 Anche con simili livelli di deficit enzimatico, i pazienti con MPS I possono manifestare un ampio spettro di sintomi e variabili livelli di gravità.1,2
Leggi anche:
Bibliografia
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001:3421-52.
- Muenzer J et al. Pediatrics 2009;123:19-29.
- Clarke LA. Expert Rev Mol Med. 2008;10:e1.
- Beck M et al. Genet Med. 2014;16(10):759-765.
- Giugliani et al. Genet Mol Biol. 2010;33(4):589-604.
- Arn P et al. J Pediatr. 2009; 154:859-64 e3.
Codice deposito aziendale MAT-IT-2100680