- Article
- Source : Campus Sanofi
- 31 oct. 2024
L’ASMD expliqué en 4 minutes
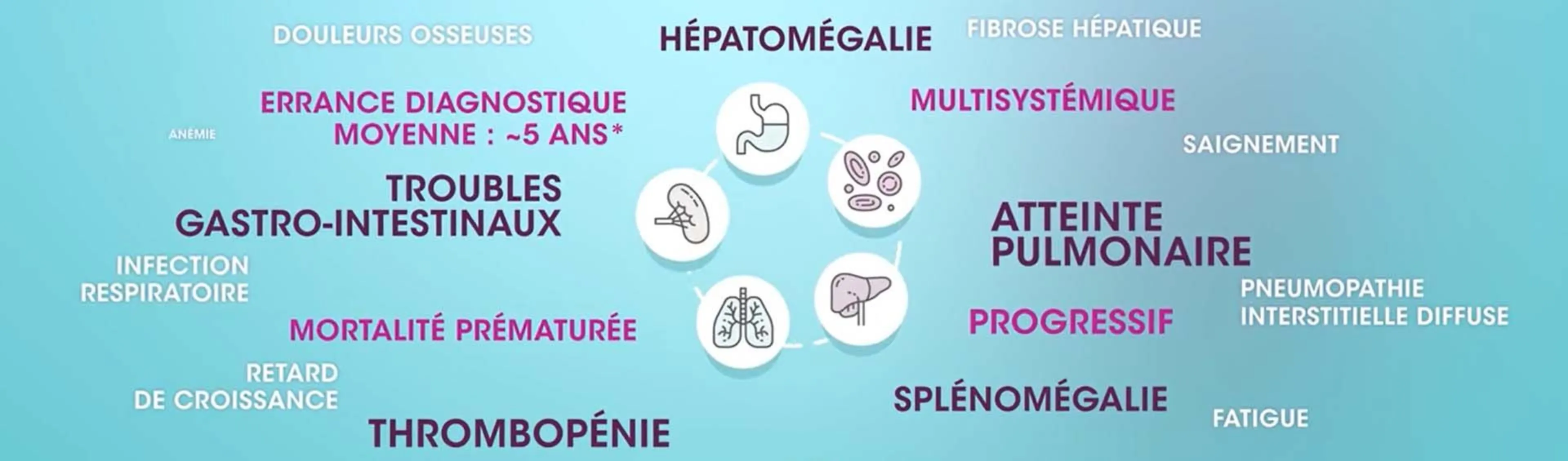
Quelle est la cause des atteintes multi systémiques de votre patient ?
Le déficit en sphingomyélinase acide ou ASMD, également connu sous le nom de maladie de Niemann-Pick de type A, A/B ou B, est une maladie génétique évolutive grave avec une morbidité importante et une mortalité prématurée.
Découvrez cette courte vidéo pour mieux comprendre l'ASMD.
Quelle est la cause des atteintes multi-systémiques de votre patient ?
Le déficit en sphingomyélinase acide ou ASMD, également connu sous le nom de maladie de Niemann-Pick de type A, A/B ou B, est une maladie génétique évolutive grave avec une morbidité importante et une mortalité prématurée.
L’ASMD est une maladie de surcharge lysosomale, causée par un déficit en sphingomyélinase acide. La sphingomyélinase acide dégrade normalement la sphingomyéline en ses composants la phosphocholine et le céramide.
Dans l’ASMD les variants pathogènes du gène SMPD1 entraînent une diminution de l'activité de la sphingomyélinase acide et donc une accumulation de sphingomyéline dans les lysosomes.
Les complications liées à l'accumulation de sphingomyéline dans les macrophages monocytes peuvent s'exprimer par des atteintes multi systémiques.
En apprenant à reconnaître les signes d'appel, vous pouvez jouer un rôle essentiel pour établir un diagnostic précoce.
Il existe un continuum d'expression clinique, on distingue trois types d’ASMD. Ils sont classés en fonction de l'âge d'apparition des symptômes, de la présence ou non d'une atteinte neurologique et de la rapidité de progression de la maladie.
L’âge d'apparition et la gravité des symptômes varient dans chaque type, mais pour tous, plusieurs organes sont touchés avec des atteintes progressives qui limitent souvent l'espérance de vie.
L’ASMD présente une grande variété de symptômes, les manifestations cliniques chez les enfants et les adultes impliquent le plus souvent des atteintes hépatiques, pulmonaires, spléniques, gastrointestinales, hématologiques. Certains types d’ASMD affectent également le système neurologique.
Les signes d'appel les plus courants sont : la splénomégalie, l’hépatomégalie, la pneumopathie interstitielle diffuse, la thrombopénie et les troubles gastro-intestinaux.
La splénomégalie et l’hépatomégalie étant souvent les premiers signes cliniques, de nombreux signes d'appel de l’ASMD sont observés également dans d'autres pathologies telles que les maladies métaboliques, les hémopathies malignes, les maladies respiratoires, cardiaques et hépatiques, pour n’en nommer que quelques-unes.
Cela rend le diagnostic d’ASMD difficile conduisant souvent à des erreurs de diagnostic. L’errance diagnostique peut aller jusqu'à cinq ans en moyenne.
Parmi les diagnostics différentiels de l’ASMD on retrouve la maladie de Gaucher, une autre maladie de surcharge lysosomale.
La maladie de Gaucher présente des signes d'appel communs avec l’ASMD, pour cette raison il est recommandé de dépister ces deux maladies en parallèle.
Le diagnostic de certitude repose sur la mesure de l'activité enzymatique. C'est un simple test sanguin. Les analyses génétiques peuvent également apporter une confirmation supplémentaire.
Le diagnostic précoce est essentiel. En reconnaissant les signes d'appel vous pouvez jouer un rôle clé pour réduire l’errance diagnostique.
Devant l'association inhabituelle de ces signes chez un de vos patients, demandez une mesure des activités enzymatiques pour écarter ces deux maladies.
Références
- McGovern MM, Avetisyan R, Sanson B-J, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD) Orphanet J Rare Diseases. 2017;12(41):1-13.2. Schuchman E, Desnick RJ.Types A and B Niemann-Pick disease. Molec Genet Metab.2017;12(41):1-13.
- Schumchman E, Desnick RJ. Types A and B Niemann-Pick disease. Molec Genet Metab. 2017;120:27-33.
- McGovern MM, Dionisi-Vici C, Guigliani R, et al. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med. 2017;19(9):967-974.
- McGovern MM, Wasserstein MP, Guiglinani R, et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick B disease. Pediatrics. 2008;122(2):e341-e349.
- Baris HN, Cohen IJ. Mistry PK. Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr Endocrinol Rev.2014;(01) :72-81.
7000048027 - 10-2024