- Article
- Source : Campus Sanofi
- 13 févr. 2026
Splénomégalie pédiatrique : quand évoquer une maladie rare ?
Extrait de la websérie 15 Min Chrono Maladies Rares, réalisée par Axis TV (Pédiatrie Pratique) avec le soutien institutionnel de Sanofi.
Intervenants :
Avec la participation de Samia PICHARD, pédiatre, Hôpital Necker, Paris
Animé par Gérard LAMBERT, Axis TV, Paris
Au sommaire :
| 00:15 | Introduction et présentation de la série « 15 Min Chrono Maladies Rares » avec la Dre Samia Pichard |
| 01:31 | Qu’est-ce qu’une splénomégalie ? Cas clinique à l’appui |
| 02:10 | Premiers examens de débrouillage |
| 03:01 | Splénomégalie isolée : premières hypothèses |
| 03:53 | Splénomégalie avec fièvre et syndrome inflammatoire |
| 04:18 | Splénomégalie avec cytopénie |
| 04:59 | Splénomégalie avec signes systémiques : et si c'était une maladie rare ? |
| 06:17 | Focus sur les maladies de surchage lysosomale |
| 08:20 | La maladie de Gaucher : points clés |
| 10:01 | La maladie de Niemann-Pick (types A, B, C) ou ASMD : points clés |
| 12:18 | Retard diagnostique et orientation vers les centres de référence |
Une rate qu'on palpe cliniquement est une rate pathologique.
Hôpital Necker, Paris
- Bonjour, bienvenue dans 15 Min Chrono Maladies Rares, une web série réalisée par Axis TV avec le soutien institutionnel des laboratoires Sanofi, et une web série consacrée à la démarche diagnostique conduisant à évoquer certaines maladies rares devant des cas cliniques complexes en pédiatrie.
L'objectif n'est pas de faire de vous des spécialistes des maladies rares ou au moins de certaines d'entre elles tant elles sont nombreuses, mais bien de savoir quand y penser devant des tableaux cliniques évocateurs afin d'éviter des retards diagnostics qui sont malheureusement trop fréquents et toujours préjudiciables pour les jeunes patients.
Et pour ce premier épisode de la saison un, j'ai le plaisir de recevoir Samia Pichard.
- Bonjour.
- Bonjour, vous êtes pédiatre, spécialiste des maladies métaboliques à l'hôpital Necker de Paris, et vous êtes également en pédiatrie générale à l'hôpital d'Argenteuil. Aujourd'hui, on va voir avec vous la conduite à tenir, la démarche diagnostique chez un enfant qui présente une splénomégalie. Vous allez nous montrer ça, ça a l'air simple mais ça ne l’est pas toujours autant que ça. Il faut des spécialistes comme vous, on va y aller tout de suite.
- Très bien.
- Alors Samia Pichard, on va partir d'un cas clinique générique de la découverte d'une splénomégalie chez un enfant. Première question, à partir de quel moment on parle de splénomégalie à l'examen clinique ?
- La splénomégalie est un symptôme purement clinique, c'est-à-dire qu'une rate qu'on palpe cliniquement est une rate pathologique. Après, il y a des variations de la normale, et il faut savoir que dans 5% des cas, on peut sentir une splénomégalie de manière tout à fait physiologique. Donc le premier élément, le premier examen paraclinique pour confirmer une splénomégalie qu'on sent sur le plan clinique et une échographie abdominale qui permettra du coup de mesurer la taille de la splénomégalie.
- Quel premier examen de débrouillage demander pour essayer de s'orienter un peu ?
- D'abord, l'anamnèse. Une anamnèse très précise, comme pour n'importe quel symptôme, bien préciser les antécédents familiaux, s'il existe une consanguinité, s'il y a des maladies génétiques, notamment dans la famille.
Et puis, surtout, les circonstances qui entourent cette splénomégalie. Y a-t-il de la fièvre ? Y a-t-il d'autres éléments extra abdominaux qui peuvent justement expliquer un petit peu ces symptômes-là ? Sur le plan biologique, il y a également des examens de débrouillage qui permettent déjà principalement d'orienter cette splénomégalie. Un bilan biologique de base associant surtout une numération formule sanguine avec un frottis sanguin, un bilan hépatique, un bilan d'hémostase, permettent déjà d'orienter la démarche diagnostique.
- Alors si l’examen clinique est normal en dehors de cette splénomégalie, on parle à ce moment-là de splénomégalie isolée. À quoi faut-il penser ?
- Quand on a une splénomégalie isolée on peut s'orienter en faisant une échographie doppler qui permettrait à ce moment-là de dire s'il existe des signes d'HTAP ou pas. Soit il existe des signes d'HTAP et donc on parle d'hypertension portale et donc là on ouvre le grand tiroir des hypertensions portales ou alors il peut s'agir de tumeurs malignes comme le cavernome portal, l'hémangiome kystique de la rate, ou alors des tumeurs malignes s'agissant d'un lymphome, par exemple.
- Alors on va voir maintenant un certain nombre de circonstances où il y a d'autres signes associés. Premiers signes associés, alors là, vous les mettez tous sur la diapositive. Lorsqu'il y a fièvre et syndrome inflammatoire, on recherche quoi ?
- On ouvre le tiroir des maladies infectieuses, il peut s'agir de maladies virales, il peut s'agir de maladies parasitaires ou alors bactériennes qu'il faudra effectivement orienter par rapport à l'anamnèse également. Et puis des examens paracliniques de base.
- Et on voit que y a un certain nombre d'étiologies bactériennes, et cetera, comme vous l'avez précisé, à rechercher. Alors maintenant on va voir une deuxième situation, c'est lorsqu'il y a une cytopénie associée.
- À ce moment-là on ouvre le tiroir des maladies hématologiques. Il peut s'agir de maladies hémolytiques constitutionnelles, soit les maladies corpusculaires, les sphérocytoses, le déficit en G6PD ou alors les hémoglobinopathies. À ce moment-là on parle de la drépanocytose, il peut également s'agir de thalassémie.
Un autre registre, on parle des anémies hémolytiques auto immunes lorsqu'on a par exemple un Coombs positif. Et finalement les hémopathies malignes qu'il faut évidemment évoquer en premier lieu, d'autant plus quand on a une clinique assez évocatrice. A ce moment-là, il s'agit des leucémies aiguës, des lymphomes, des leucémies myéloblastiques.
- Alors après, lorsqu'on a des signes maintenant un petit peu généraux, une dysmorphie, un syndrome neurologique, un retard statural. Donc là on a affaire à des maladies un peu systémiques ?
- Alors là, on ouvre effectivement le tiroir des maladies rares, de ce qu'on appelle les maladies de surcharge lysosomale, qu'on peut différencier en parlant des sphingolipidoses, à ce moment-là qu’on reconnaît la maladie de Gaucher, le déficit en sphingomyélinase. Il existe également des mucopolysaccharidoses lorsqu'on a une dysmorphie assez spécifique qui peut orienter vers ces maladies-là, ou alors quand on a des signes neurologiques, et là à ce moment-là, on parle des neurolipidoses ou des lipidoses : on parle de maladie de Niemann-Pick C, on parle de maladie de Farber par exemple.
- Alors on va préciser un petit peu ces maladies, mais je voulais vous demander dans le dernier cas, lorsqu'il y a hypersplénisme et hépatopathie.
- À ce moment-là, on revient à la première diapositive lorsqu'on parle de maladie portale, on fait l'échographie doppler pour s'orienter. Soit il s'agit d'une hypertension portale ou alors on parlera plutôt de maladie spécifique de la rate, et à ce moment-là ce sont soit des tumeurs malignes ou des tumeurs non malignes que je vous avais expliquées à la première diapo.
- Merci pour cette démarche diagnostique.
Alors Samia Pichard, on a vu que lorsque certains signes sont associés, en plus de l'aspect splénomégalie hein, on a parlé de dysmorphie, de signes neurologiques, de retard statural. Il faut penser à des maladies lysosomiales, de surcharge lysosomiale. De quoi parle-t-on ? Est-ce que vous pouvez nous préciser ?
- Tout à fait. Alors le lysosome - je vais vous parler un petit peu de biochimie et de cette petite organelle qui existe à l'intérieur de toutes cellules et qui permet en gros de dégrader les macromolécules, grâce à différentes hydrolases en milieu acide. Les macromolécules vont rentrer à l'intérieur du lysosome, vont se dégrader pour pouvoir sortir du lysosome. En pathologie, Il existe un déficit en général d'une enzyme ou d'un transporteur qui fait qu'il va y avoir une surcharge à l'intérieur de ce lysosome. Il va en découler une ballonnisation et donc une dégradation de la cellule où se trouve cette accumulation de produits. De ce fait, on peut avoir différents signes cliniques qui sont principalement progressifs. C'est à dire que les enfants vont bien en général à la naissance et petit à petit, il va y avoir des symptômes cliniques qui vont arriver au fur et à mesure de la vie de l'enfant. Et il s'agit surtout de maladies plurisystémiques parce que le lysosome, on en a dans toutes les cellules et il va y avoir une accumulation progressive. Il peut y avoir des atteintes neurologiques, des atteintes hématologiques avec une accumulation au niveau du foie, de la rate, une hépatosplénomégalie, des manifestations effectivement également au niveau de l'œil, du cœur. Tous les organes finalement peuvent être atteints dans ces maladies-là.
On les classifie principalement en fonction de la surcharge de la molécule qui va être surchargée. Quand ce sont des sphingolipides, on va avoir des sphingolipidoses, quand ce sont des Mucopolysaccharides, ça va être des mucopolysaccharidoses, et cetera. Donc on peut décrire un certain nombre de maladies. Il y en a plus d'une cinquantaine de maladies lysosomales et dont la clinique est quasiment toute commune avec cette association de maladies plurisystémiques et de maladies très progressives.
- Alors parmi celles-ci, il y a notamment la maladie de Gaucher par exemple.
- Tout à fait. Alors la maladie de Gaucher, effectivement, a été décrite en 1880 par Monsieur Ernest Gaucher qui avait décrit un patient de 60 ans qui avait une grosse rate. Et en fait, ce n'est que beaucoup plus tard, en 1960, qu'on a compris que cette maladie était liée à un déficit enzymatique, le déficit de la bêta-glucocérébrosidase qui était lié au gène GBA qu'on a découvert que beaucoup plus tard. Et en fait cette maladie est autosomique récessive. Maintenant on le sait avec un type 1 qui est beaucoup plus fréquent, qu'on retrouve dans plus de 94% des cas. Il s'agit d'une maladie rare. Il y a un registre national de la maladie de Gaucher en France qui a pu recenser 500 patients en 30 ans de 1980 à 2010, et certains patients sont traités. Il y a 6000 patients traités à ce jour. Il s'agit du coup d'un déficit de la glucocérébrosidase, cette enzyme qui va finalement avoir comme conséquence une accumulation du glucosylcéramide et du glucosylsphingosine, qui vont s'accumuler au niveau des macrophages, d'où l'hépatosplénomégalie qui est le principal symptôme de cette maladie-là.
Comme vous le voyez sur cette diapo, le type 1 est le plus fréquent avec plus de 90% de patients malades. Le type 3 concerne les formes neurologiques où il peut y avoir une atteinte neurologique associée à l'atteinte viscérale. Et dans le type 2, ce sont des formes beaucoup plus sévères. Des formes principalement de l'enfant, qui souvent décèdent avant l'âge de 2 ans, qui heureusement, sont beaucoup, beaucoup plus rares.
- Alors une autre maladie qui portait un autre nom auparavant : on parlait de la maladie de Niemann-Pick, on a encore ce terme en en tête.
-Bah oui, Niemann-Pick parce que c'est Monsieur Niemann qui l'avait décrit en 1914 quand il avait décrit un bébé qui avait un gros foie, une grosse rate et qui a eu par la suite une régression psychomotrice. Et puis Monsieur Pick, beaucoup plus tard dans les années 30, qui a décrit sur le plan anatomopathologique la surcharge en lipides de cette maladie-là. Et pareil c'est qu'en 1960 où Monsieur Rasco Brady a finalement décrit ce déficit de l'enzyme qui est le déficit en sphingomyélinase, et du coup qui décrit la maladie de Niemann-Pick de type AB, à ne pas confondre avec la maladie de Niemann-Pick de type C qui est un autre type de maladie de surcharge lysosomale également.
Donc, là aussi, il va y avoir un déficit d'une enzyme, le déficit en sphingomyélinase, avec une accumulation de sphingomyéline en amont qui va principalement s'accumuler au niveau du foie, au niveau de la rate, d'où l'hépatosplénomégalie. Mais la différence avec la maladie de Gaucher, c'est qu'il y a également une atteinte au niveau des poumons. Et ce sont des patients qui vont avoir d'autres symptômes cliniques, notamment des symptômes pulmonaires, avec une atteinte plutôt de type interstitiel. Dans les formes plus sévères, les formes A, les formes AB, il peut y avoir également une atteinte neurologique.
Il est également décrit dans ces maladies-là un retard statural et également un retard pubertaire qui peut survenir dans ces maladies-là.
On parle de maladie de Niemann-Pick A et de maladie de Niemann-Pick B mais en fait, il existe un spectre phénotypique entre le type A et le type B avec une forme très sévère qui est le type A où il va y avoir une atteinte neurologique rapidement progressive et l'atteinte de type B qui est souvent une maladie de l'adulte, avec des patients qui ont une hépatosplénomégalie avec une atteinte interstitielle pulmonaire.
Et puis, entre les 2 formes, on peut avoir des formes de pathologies de patients qui ont une atteinte interstitielle et qui peuvent avoir une atteinte neurologique a minima. C'est ce qu'on appelle la forme de Niemann-Pick AB.
-Alors, Samia Pichard, à travers ce cas clinique d'un enfant présentant une splénomégalie, on a vu à quel moment il faut suspecter une maladie de surcharge lysosomiale. Il y a un problème aujourd'hui, c'est que ces maladies sont souvent diagnostiquées tardivement.
-Tout à fait. Il y a un retard malheureusement diagnostic assez important dans ces maladies-là vu leur rareté.
Dans la maladie de Gaucher, on est autour de 5 ans de retard diagnostic. Dans la maladie de Niemann-Pick AB, on est plus autour de 7 ans de retard de diagnostic dans ces maladies-là.
-À votre avis, comment on pourrait améliorer ce délai ? Parce que finalement il est toujours préjudiciable pour les enfants.
-Donc oui, effectivement, il faudrait essayer de sensibiliser au maximum les médecins, les médecins généralistes, les pédiatres mais également les médecins spécialistes d'organes qui pourront plus facilement poser le diagnostic de ces maladies-là.
- Avec finalement comme message quand y penser et quand adresser dans un centre de référence par exemple.
-Tout à fait.
-Adresser à vous.
-Ou à mes collègues.
-Notamment. Ou à vos collègues. Samia Pichard, merci.
-Merci à vous.
Gaucher ou ASMD ? Consultez cet algorithme pratique pour explorer la splénomégalie pédiatrique et orienter le diagnostic étape par étape :
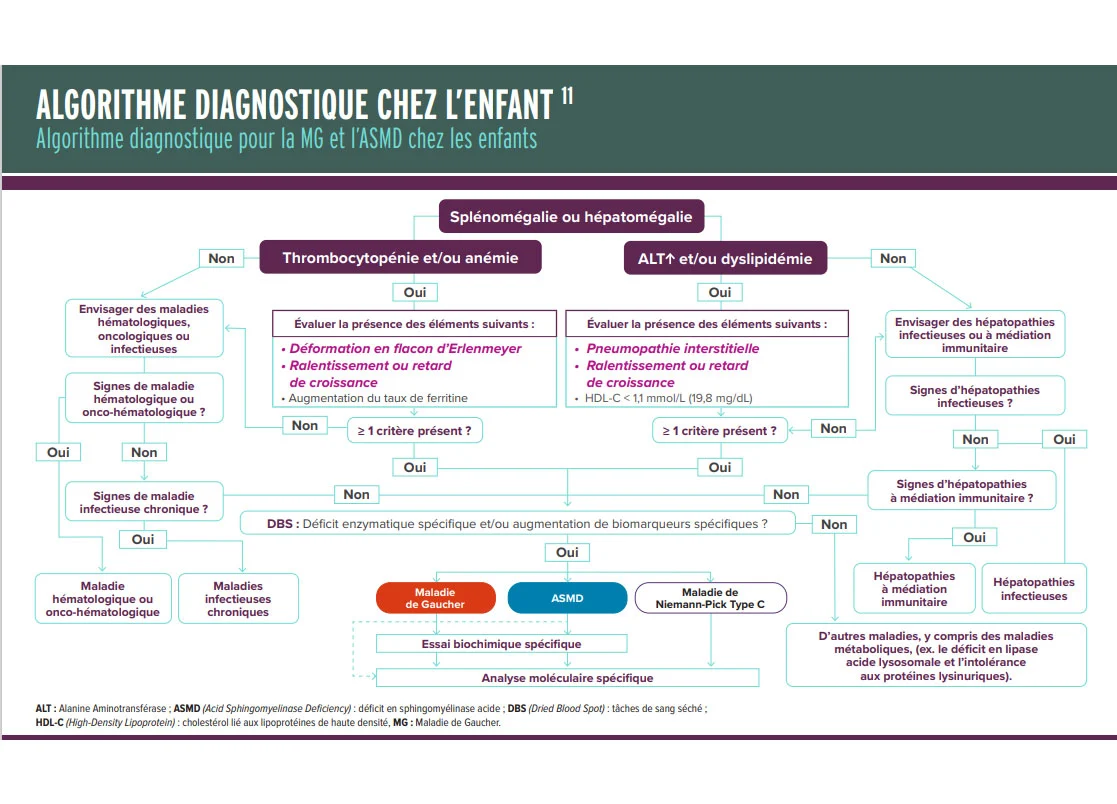
Documents d’intérêts sur l’ASMD : fiche synthèse et algorithmes diagnostiques adulte et pédiatriques
Fiches d’intérêts concernant l’ASMD: définition, présentation clinique, symptômes orientant le diagnostic et diagnostic de certitude, algorithmes diagnostiques adulte et pédiatrique mettant en avant l’importance du parallel testing entre la Maladie de Gaucher et l’ASMD.
MAT-FR-2504330 - 03/2026