- Articolo
- Fonte: Campus Sanofi
- 28 apr 2024
Aspetti genetici

La trasmissione della Malattia di Fabry ha carattere ereditario e, più precisamente, è legata al cromosoma X (X-linked). Il difetto genetico legato a tale patologia, che comprende oltre 640 mutazioni del gene GLA1, può essere pertanto trasmesso da entrambi i sessi.
Più precisamente:
- Gli uomini portatori del gene mutato, lo trasmetteranno a tutte le figlie femmine, ma mai ai figli maschi.
- Le donne portatrici hanno una probabilità pari al 50% di trasmetterlo alla progenie, indipendentemente dal sesso del nascituro.
Trasmissione ereditaria della mutazione GLA (padre)
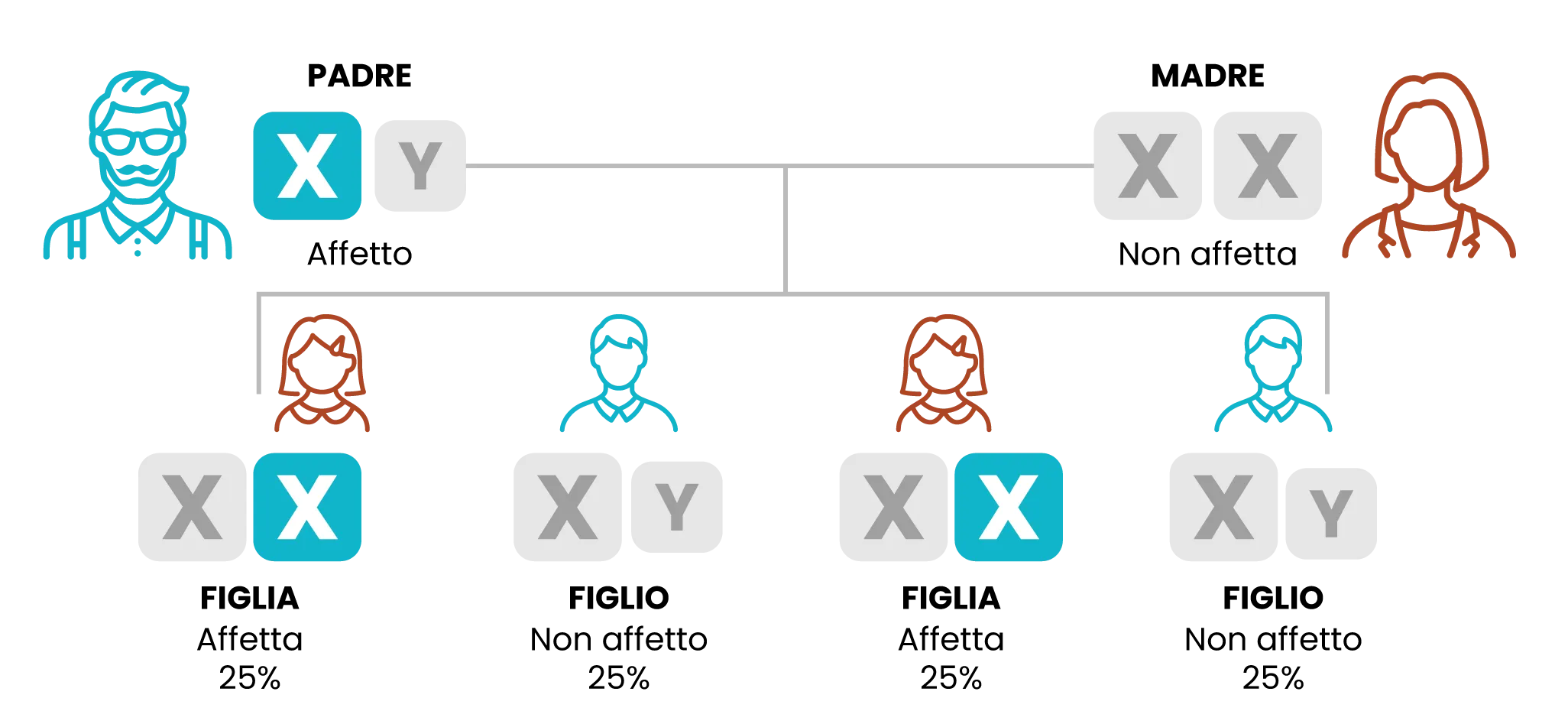
I maschi con il gene difettoso lo trasmettono a tutte le figlie e a nessuno dei figli.
Padre affetto da definirsi come emizigote con il gene difettoso/la mutazione e femmine affette come eterozigoti con il gene difettoso.
Trasmissione ereditaria della mutazione GLA (madre)
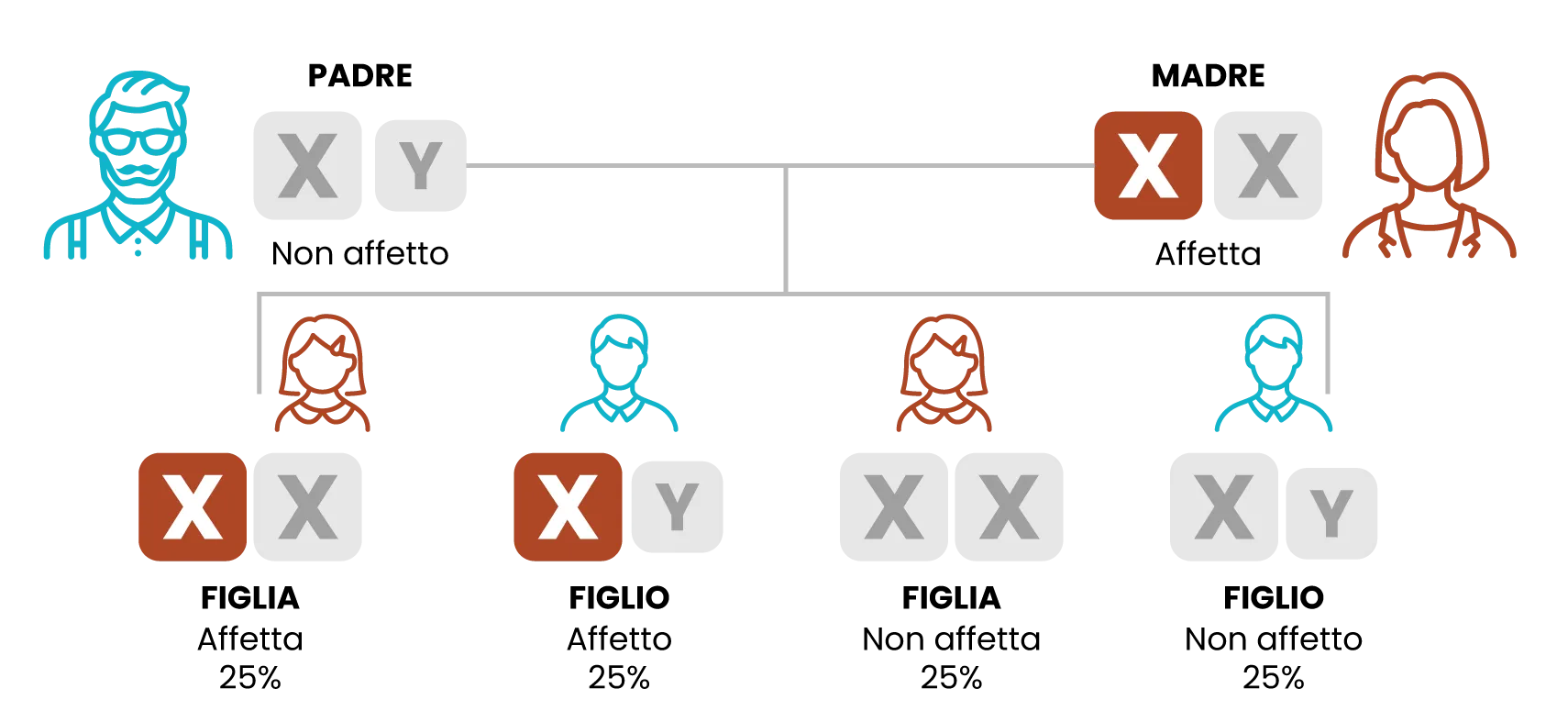
Le donne con il gene difettoso hanno una probabilità del 50% di trasmetterlo durante ogni gravidanza.
Padre affetto da definirsi come emizigote con il gene difettoso/la mutazione e femmine affette come eterozigoti con il gene difettoso.
Nonostante l'anamnesi familiare sia un indubbio indicatore per la diagnosi di Malattia di Fabry, la maggior parte delle mutazioni è ancora non classificata e vi sono troppo poche informazioni sulla storia naturale per trarre conclusioni. Sono state dunque identificate diverse mutazioni spontanee o ex novo.2,3
Le mutazioni note, invece, possono essere classificate nelle categorie Malattia di Fabry classica, Malattia di Fabry non classica, polimorfismi benigni o pseudodeficienze.1 È possibile osservare un’ampia variabilità fenotipica in termini di età di esordio, velocità di progressione, coinvolgimento d’organo e gravità.
La Malattia di Fabry nelle donne
La Malattia di Fabry nel sesso femminile merita un cenno a parte: un tempo ritenute asintomatiche, le donne possono in realtà sviluppare manifestazioni cliniche di entità variabile, da lieve a grave.4–7
L’analisi delle caratteristiche cliniche basali di una coorte di 1765 maschi e femmine nel Registro Fabry ha dimostrato che l’età mediana di esordio dei sintomi era 13 anni nel sesso femminile, rispetto a 9 anni nel sesso maschile.7 Nelle femmine, il dolore neurologico era il sintomo di presentazione più comune, riportato nel 41% delle pazienti, e l’età mediana di esordio era 10 anni. Per altri sintomi, l’età di esordio era 12-32 anni, rispetto a 8-20 anni nei maschi.
Si ritiene che l’espressione variabile dei sintomi nelle femmine sia influenzata dall’inattivazione del cromosoma X, un fenomeno in cui una delle due serie aploidi dei geni X-linked in ciascuna cellula viene inattivata e non presenta alcuna espressione fenotipica. Un’analisi ha dimostrato che l’inattivazione del cromosoma X rappresenta un fattore importante nel determinare la gravità e il coinvolgimento clinico nelle femmine eterozigoti e che esiste una differenza statisticamente significativa tra i punteggi di gravità dei sintomi delle pazienti con pattern di inattivazione del cromosoma X bilanciati e sbilanciati.8 Il pattern di inattivazione può variare da un organo all’altro, pertanto una paziente di sesso femminile può manifestare sintomi gravi a carico di alcuni organi, pur restando asintomatica a livello di altri.
Leggi anche:
Bibliografia
- http://fabry-database.org/ (Ultimo access: 23/02/2020).
- Redonnet-Vernhet I et al. J Med Genet 1996;33:682–88.
- Hasholt L et al. J Med Genet 1990;27:303–306.
- Deegan PB et al. J Med Genet 2006;43:347–52.
- MacDermot KD et al. J Med Genet 2001;38:769–75.
- Wang RY et al. Genet Med 2007;9:34–45.
- Eng CM et al. J Inherit Metab Dis 2007;30:184–92.
- Dobrovolny R et al. J Mol Med. 2005;83(8):647-654.
Codice deposito aziendale MAT-IT-2100588