- Articolo
- Fonte: Campus Sanofi
- 12 mag 2024
Patogenesi della Malattia di Fabry

La Malattia di Fabry rappresenta uno di oltre cinquanta disordini ereditari rari noti, denominati disordini da accumulo lisosomiale. Ognuno di essi è causato da un difetto genetico ereditario responsabile della carenza di uno o più specifici enzimi lisosomiali.2 L’età di esordio, l’organo o gli organi coinvolti e la gravità di questi disordini mostrano una notevole variabilità, ma il decorso è sempre progressivo.
La Malattia di Fabry è causata da una mutazione nel gene GLA, che codifica per la α-galattosidasi lisosomiale (nota anche come α-GAL, α-Gal-A e ceramide triesossidasi).1 Il deficit parziale o completo di attività dell’α-GAL determina una ridotta capacità di catabolizzare i lipidi con residui terminali α-galattosil. Questi lipidi, in particolare la globotriaosilceramide (nota anche come GL-3, Gb3 e ceramide triesosside CTH (ceramide trihexoside), si accumulano nei lisosomi di vari tipi di cellule in tutto il corpo, incluse le cellule endoteliali capillari, le cellule renali, le cellule cardiache e le cellule nervose, con conseguente danno multisistemico progressivo. Nel tempo, ciò può portare a danno d’organo a carico dei reni, del cuore e/o del sistema cerebrovascolare.
Accumulo di GL-3 nell’endotelio capillare
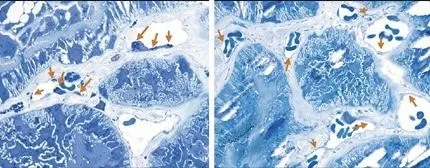
Micrografia elettronica dell’endotelio capillare renale in un paziente con Malattia di Fabry: le frecce indicano aree di accumulo di GL-3.
Immagine della galleria Genzyme utilizzata su autorizzazione.
Leggi anche:
Bibliografia
- Germain DP. Orphanet J Rare Dis. 2010 Nov 22;5:30.
- Desnick RJ, Ioannou YA, Eng CM (2014). α-Galactosidase A Deficiency: Fabry Disease. In Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis S.E., Ballabio A, Gibson K, Mitchell G (Eds).
Codice deposito aziendale MAT-IT-2100588