- Articolo
- Fonte: Campus Sanofi
- 13 mag 2024
Panoramica della Malattia di Pompe

La Malattia di Pompe è un disordine neuromuscolare, progressivo, multisistemico, debilitante, potenzialmente fatale. È stata descritta per la prima volta dal patologo olandese Joannes C. Pompe nel 1932, in una bambina di 7 mesi morta di ipertrofia cardiaca idiopatica1 in cui è stato riscontrato un massiccio accumulo di glicogeno in diversi tessuti, specialmente nei muscoli scheletrici e cardiaci.
Nel 1963 la malattia è stata correlata ad un deficit ereditario dell'enzima lisosomiale alfa-glucosidasi acida (GAA),2 responsabile della degradazione del glicogeno in glucosio. Il risultato è un accumulo intralisosomiale di glicogeno, principalmente nelle cellule muscolari, che causa la perdita progressiva della funzionalità muscolare.
Scopri gli apparati e i sistemi che possono essere colpiti dalla Malattia di Pompe
.jpg)
%20(1).jpg)
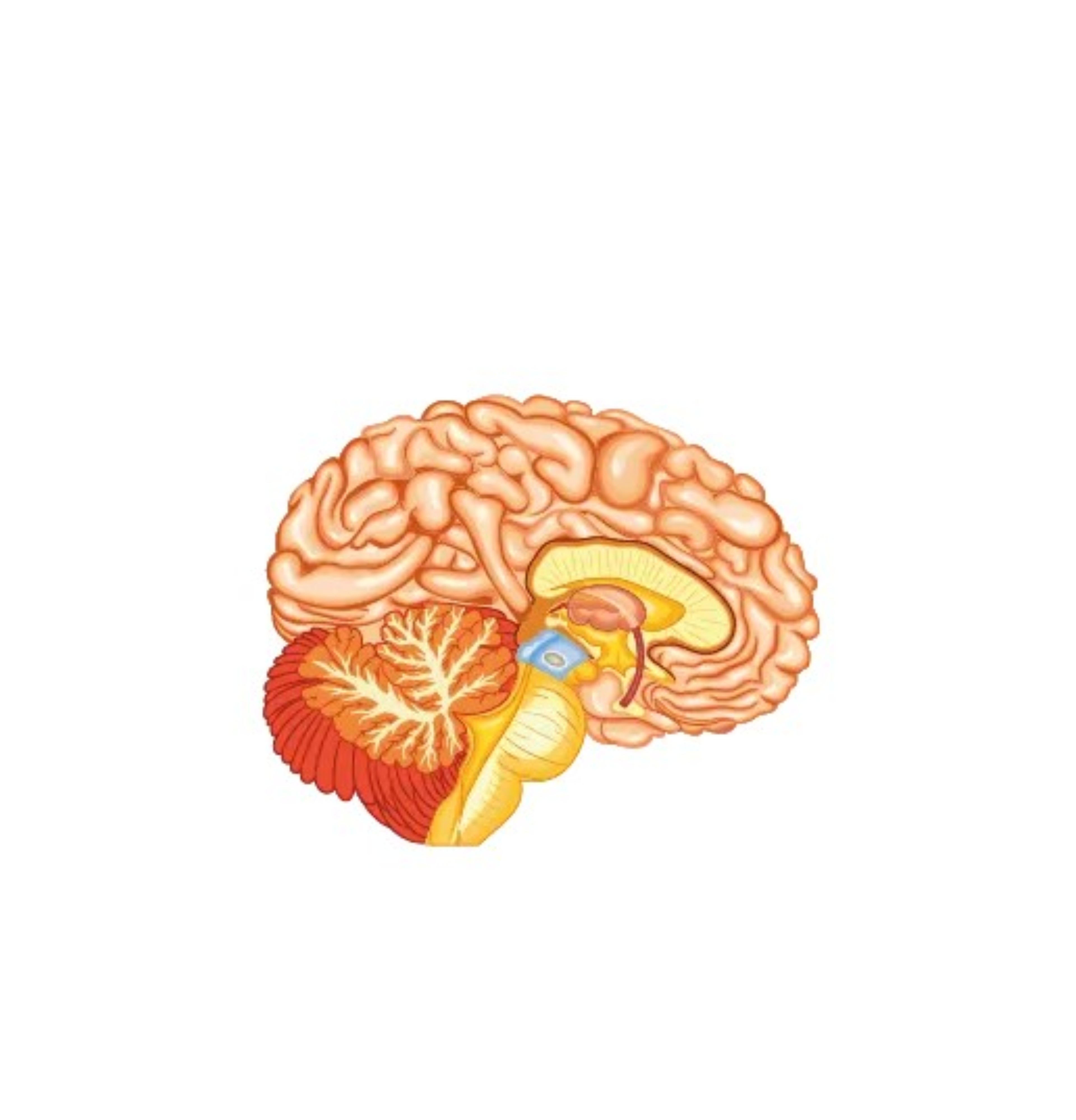
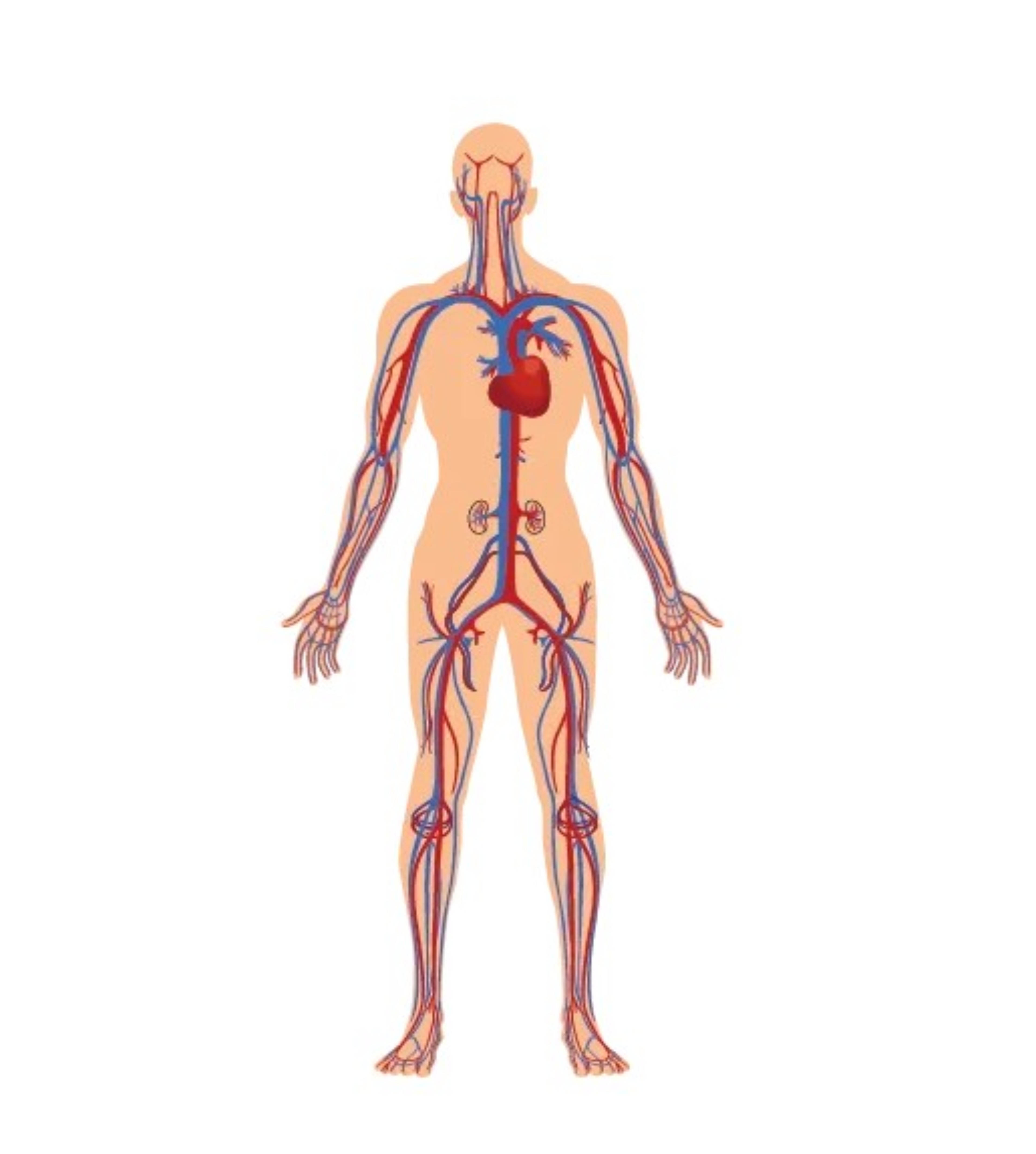
Sistema nervoso centrale
Nella Malattia di Pompe è presente un accumulo di glicogeno lisosomiale a livello di encefalo e cellule nervose del corno anteriore, dei nervi periferici e dei muscoli lisci. Sono pure presenti vacuoli PAS-positivi nelle cellule della muscolatura liscia della tunica media delle arteriole cerebrali associati anche a dilatazioni aneurismatiche di modeste dimensioni. Tale patologia, propria del sistema vascolare, può indurre la formazione di aneurismi cerebrali e di una arteriopatia dilatativa cerebrale. Nei pazienti adulti affetti da Malattia di Pompe trova indicazione l’esecuzione di angio-RM dell’encefalo al fine di reperire precocemente malformazioni vascolari cerebrali le quali potrebbero essere causa di eventi potenzialmente mortali, quali l’emorragia sub-aracnoidea e la compressione del tronco cerebrale. La medesima alterazione della struttura vascolare può essere responsabile di una ipoacusia di tipo neuro-sensoriale, conduttiva o mista.
Sistema nervoso periferico
.2024-07-11-12-31-28.jpg)
I pazienti affetti da Malattia di Pompe possono accusare parestesie, a carattere doloroso ed urente, specie a carico delle parti distali degli arti inferiori: la cosiddetta neuropatia delle piccole fibre. Risulta inoltre possibile osservare la sindrome dei piedi brucianti.
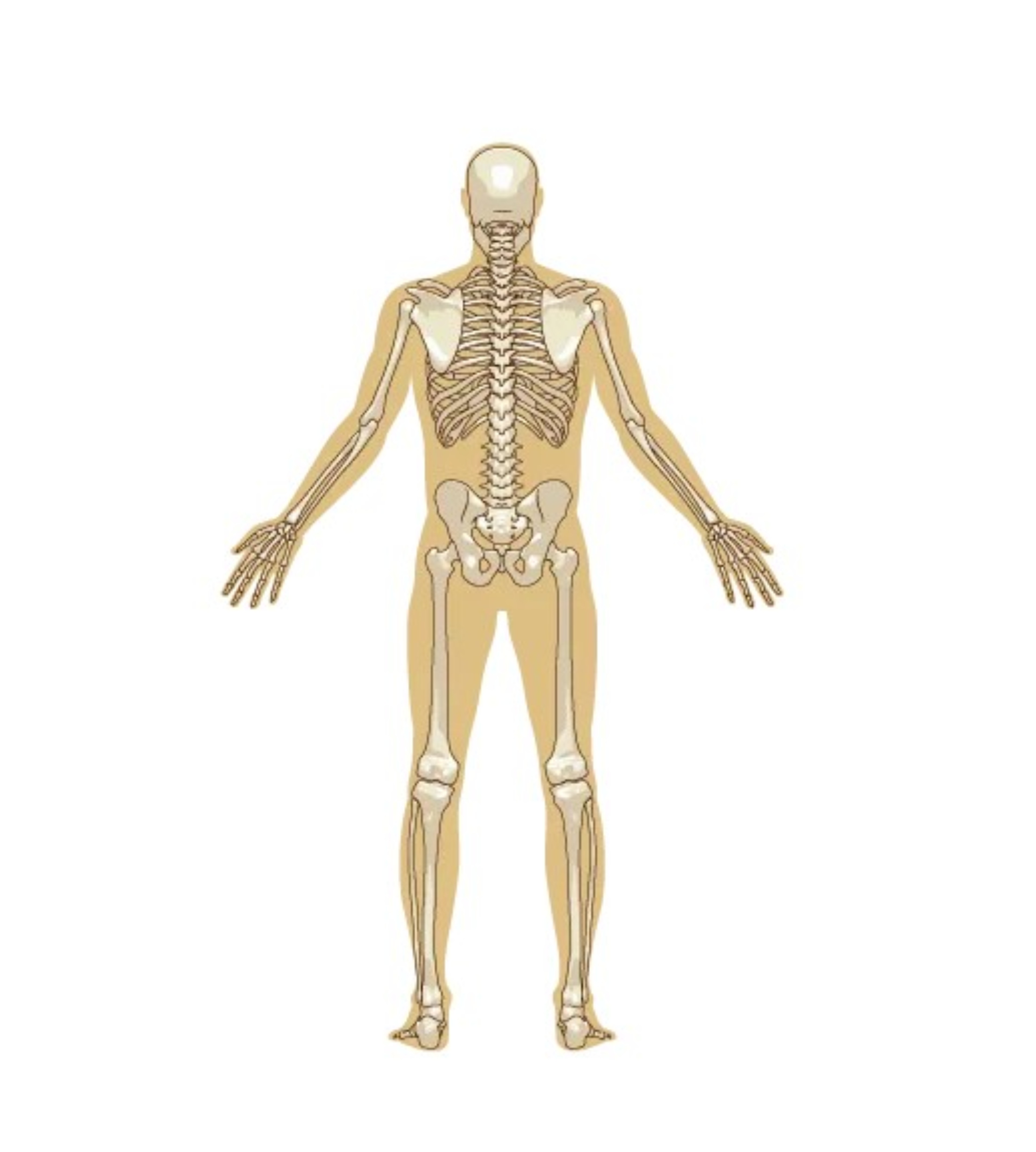
Apparato scheletrico
La riduzione della densità minerale ossea con conseguente degenerazione ossea progressiva, dovuta anche al deficit di alfa-glucosidasi, associata ad una debolezza delle strutture muscolari comporta la possibile presenza di osteoporosi, fratture ossee, scoliosi, cifosi e lordosi lombare. La cosiddetta sindrome della colonna curva, disturbo neuromuscolare caratterizzato da una limitata flessione della colonna cervicale e dorso-lombare, si associa frequentemente ad una grave insufficienza respiratoria di tipo restrittivo che può comportare anche l’utilizzo di supporti respiratori. Le fratture ossee interessano essenzialmente le ossa lunghe, quali il femore e l’omero, specie in pazienti immobili e allettati per lungo tempo.
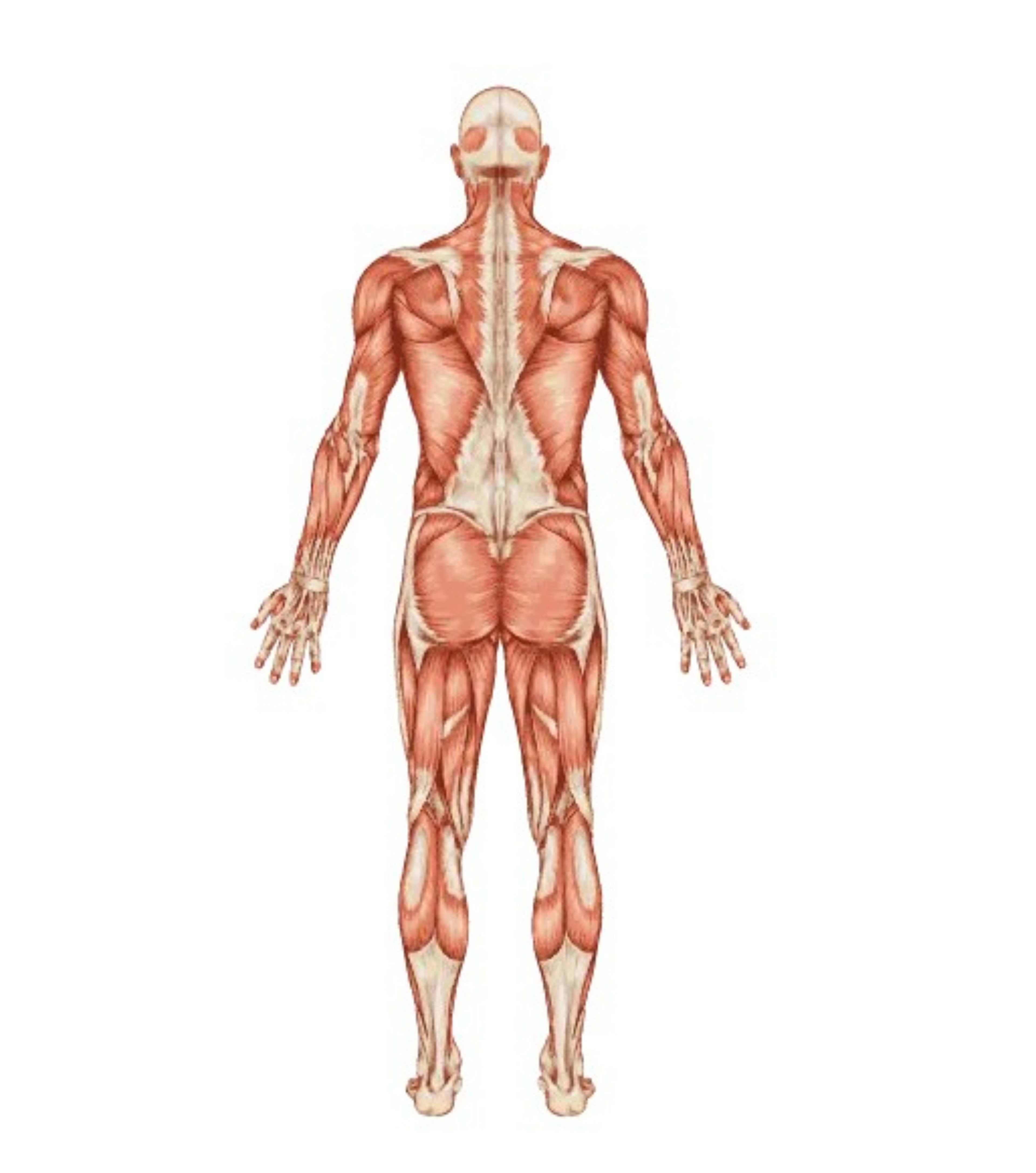
Apparato muscolare
Il deficit di alfa-glucosidasi determina un accumulo di glicogeno lisosomiale in numerosi tessuti dell’organismo, in particolare a livello di muscoli scheletrici, muscoli respiratori e cuore. I sintomi caratteristici sono rappresentati da dolori muscolari, affaticamento, intolleranza all’esercizio fisico. I pazienti lamentano difficoltà alla deambulazione e nel salire le scale, nell’alzarsi dal letto, dalla sedia, da terra. Tali sintomi trovano correlazione con un aumento della creatinfosfochinasi sierica (CPK >100) e con un incremento dei valori degli enzimi ALT (>45) e AST (>55). La ridotta funzionalità muscolare può interessare i muscoli fissatori della scapola e determinare un’alterazione di posizione della scapola, detta “scapola alata”.
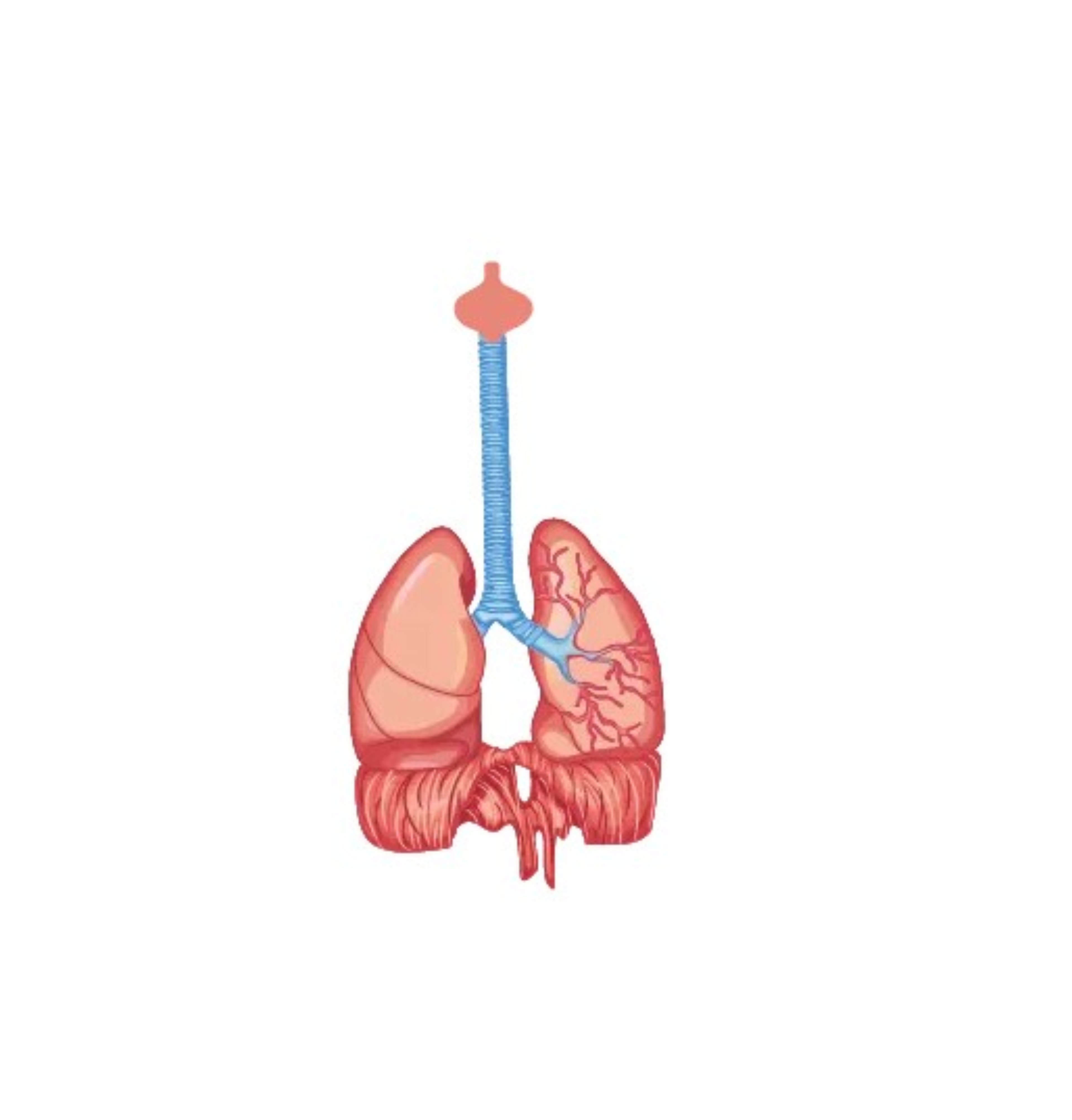
Apparato respiratorio
I sintomi respiratori iniziali sono rappresentati da disturbi respiratori del sonno con un’evoluzione verso una ipoventilazione notturna, necessitante di ventilazione con CPAP, una forma di dispnea particolarmente accentuata in posizione supina, una tosse inefficace accompagnata da insufficienza respiratoria. I disturbi del sonno determinano una riduzione della qualità di vita causando sonnolenza diurna e affaticamento. A causa della debolezza muscolare la tosse può risultare non efficace.
Apparato cardiovascolare
Nel muscolo cardiaco l’accumulo di glicogeno determina disturbi della conduzione atrio-ventricolare e una ipertrofia del miocardio. I disturbi del ritmo sono essenzialmente rappresentati da aritmia sinusale, tachicardia sopra-ventricolare, sindrome di Wolff-Parkinson-White. La parete delle arterie evidenzia la presenza di un accumulo di glicogeno nelle cellule della muscolatura liscia. Le arterie diventano rigide, sono poco elastiche e possono andare incontro a fenomeni degenerativi i quali conducono alla formazione di arteriopatia dilatativa e di aneurismi.
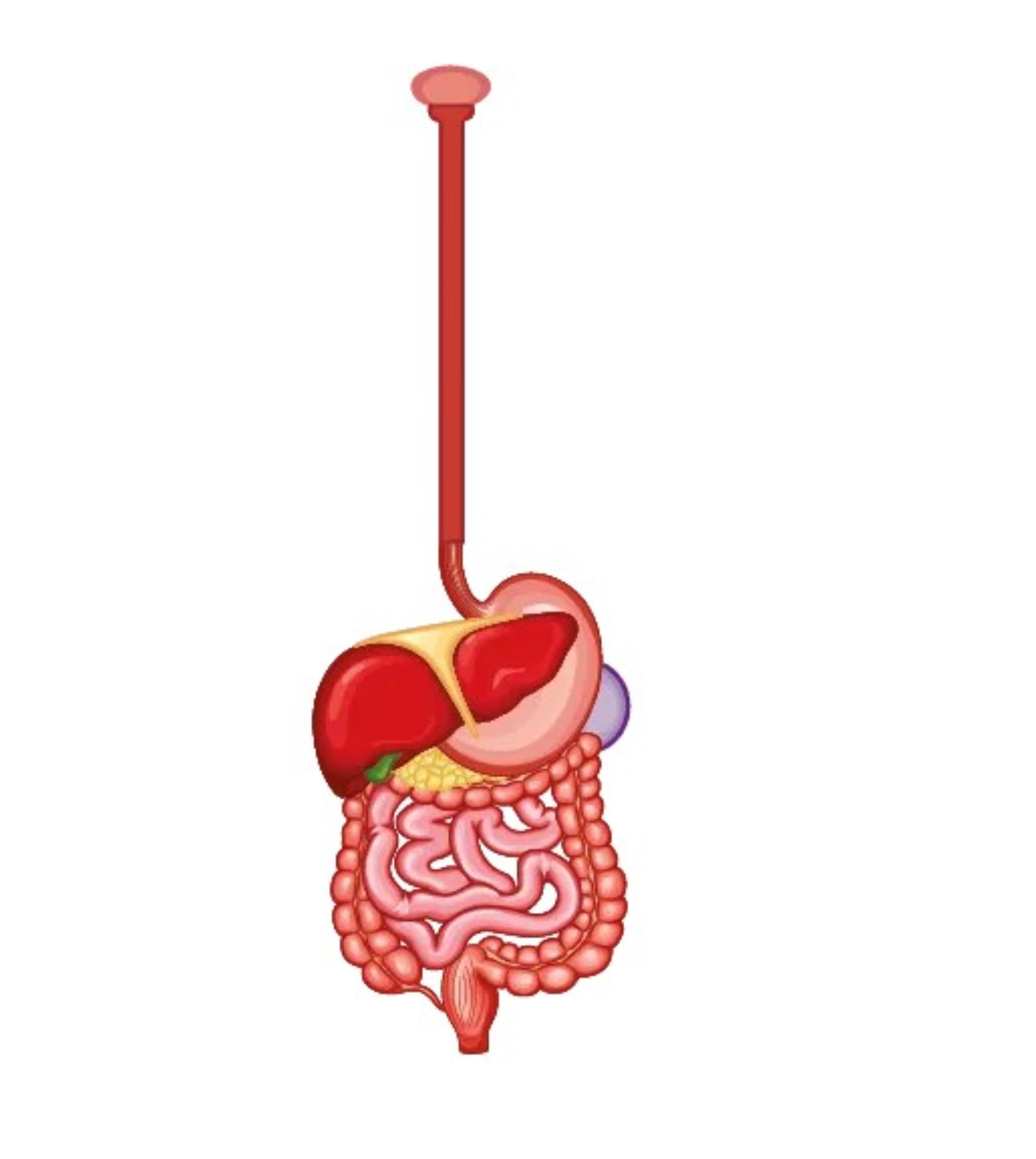
Apparato gastroenterico
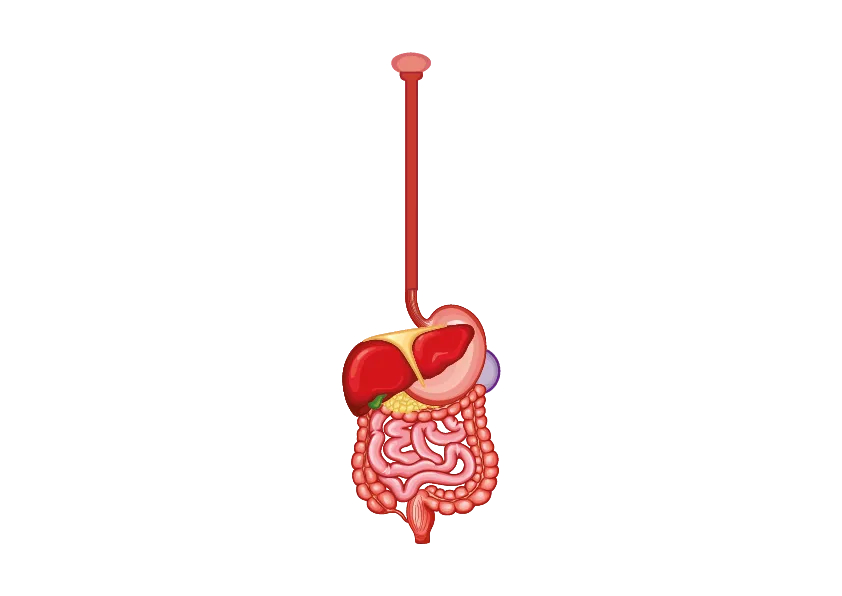
Il coinvolgimento dei muscoli bulbari può causare, oltre a disturbi della deglutizione e a disfagia, anche la disartria. L’accumulo di glicogeno nella muscolatura liscia intestinale e il danno vascolare proprio della sierosa dell’intestino può determinare una alterazione della peristalsi e quindi del transito intestinale. I sintomi gastro-enterici possono quindi essere riferibili a dolori addominali, inappetenza, sazietà precoce, vomito, diarrea cronica, incontinenza fecale.
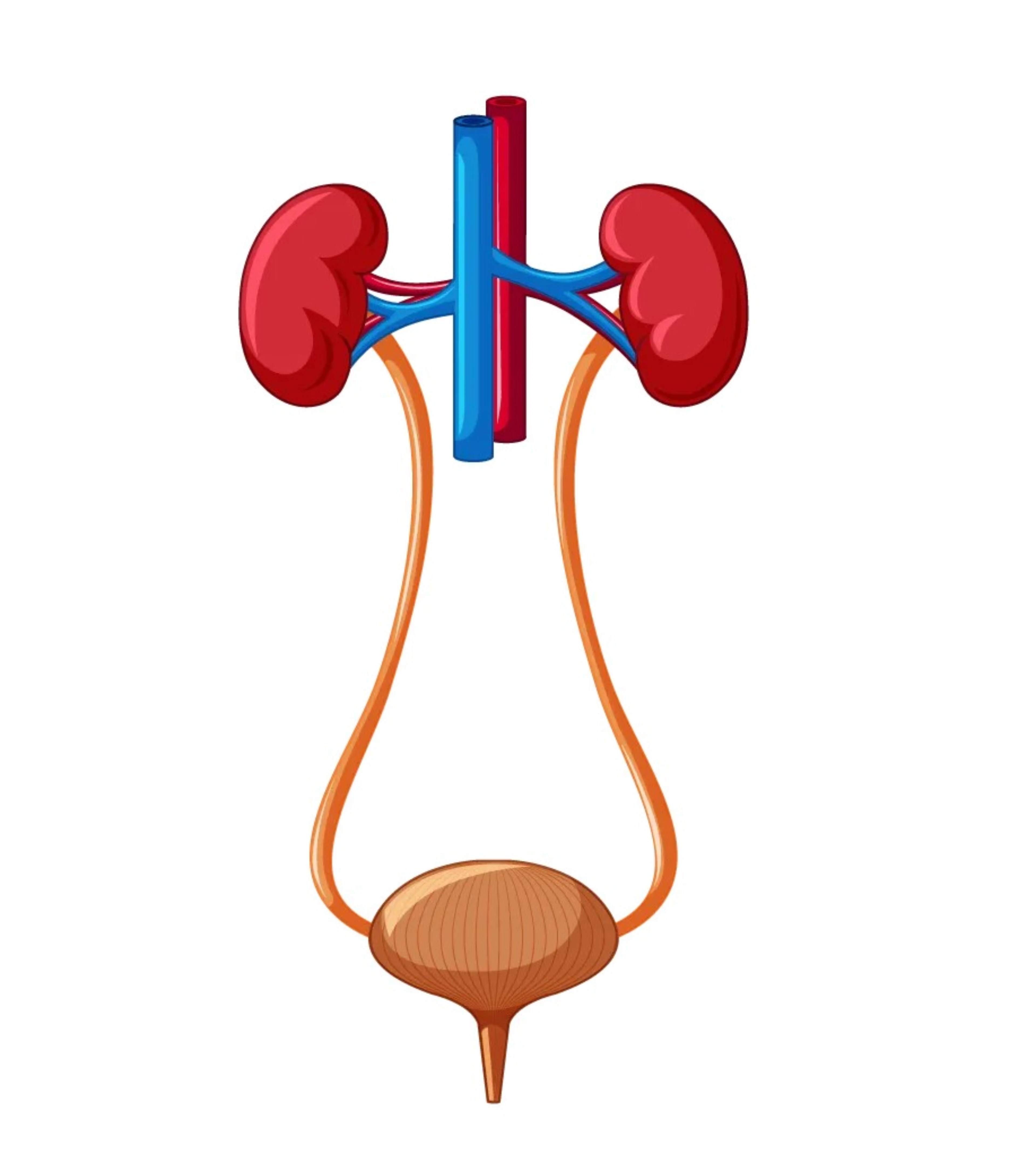
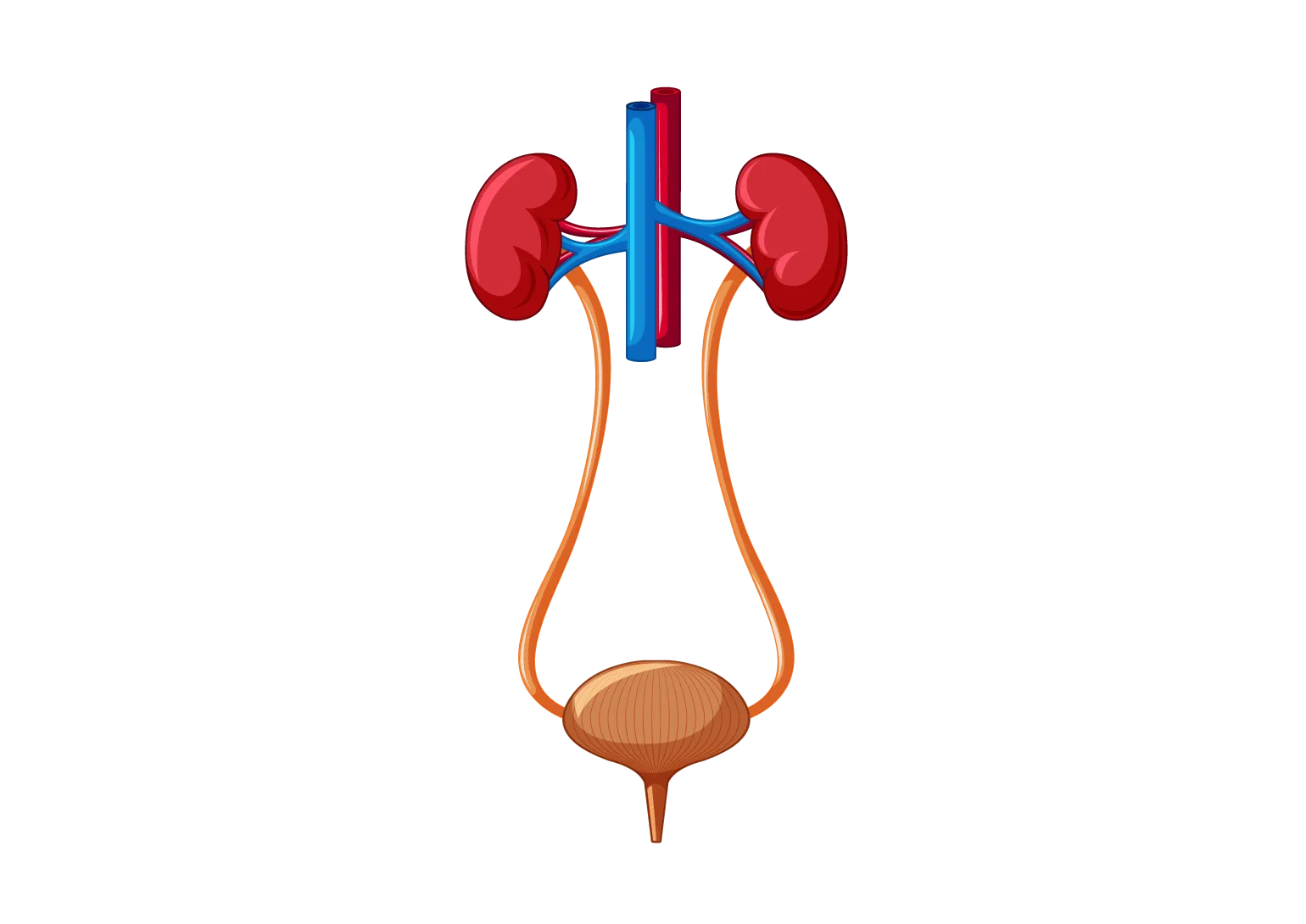
Apparato uro-genitale
%20(1).2024-01-02-07-29-09.jpg)
Le alterazioni motorie della muscolatura liscia della vescica causate da un accumulo di glicogeno possono determinare incontinenza urinaria.
Scarica il pdf con gli apparati e i sistemi che possono essere colpiti dalla Malattia di Pompe!
Leggi anche:
Bibliografia
- Pompe JC. Ned Tijdschr Geneeskd 1932; 76:304-311.
- Hirschhorn R et al. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G., eds. The Online Metabolic and Molecular Bases of Inherited Disease. OMMBID.
Codice deposito aziendale MAT-IT-2100349