- Articolo
- Fonte: Campus Sanofi
- 5 mag 2024
Patogenesi della Malattia di Pompe

Sul cromosoma 17 è situato un gene (17q25.2-q25.3) che codifica per la produzione di alfa-glucosidasi acida (GAA), un enzima responsabile della degradazione del glicogeno in glucosio all'interno dei lisosomi.1,2
Le mutazioni di questo gene causano un deficit dell'attività dell'enzima GAA, con un conseguente accumulo intralisosomiale di glicogeno, prevalentemente nelle cellule muscolari.2
Un accumulo continuo di glicogeno provoca rigonfiamento e rottura dei lisosomi, con conseguente danno cellulare. Questo, a sua volta causa una degenerazione progressiva dei muscoli scheletrici e respiratori (e del muscolo cardiaco nei neonati), con possibile perdita della funzionalità.1,3
L’immagine di micrografia elettronica di una cellula di muscolo scheletrico rappresenta un esempio di accumulo di glicogeno e della patologia muscolare conseguente, che frequentemente si manifesta prima di qualsiasi segno o sintomo clinicamente rilevabile.
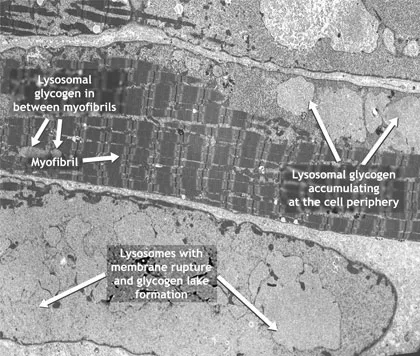
Micrografia elettronica di cellule muscolari in neonato affetto da Malattia di Pompe
L'accumulo di glicogeno causa l'ingrossamento dei lisosomi che invadono in modo anomalo lo spazio cellulare. Nonostante alcune miofibrille sane possano essere ancora presenti nello stadio iniziale della Malattia di Pompe, nello stadio avanzato sono quasi completamente sostituite dal glicogeno, con conseguente compromissione della funzionalità muscolare.
Adattato da rif.4
Il glicogeno che si forma nella Malattia di Pompe tipicamente NON causa anomalie del metabolismo del glucosio, come l'ipoglicemia, in quanto il glicogeno immagazzinato nei lisosomi non funge da riserva di glucosio.
Mentre nella Malattia di Pompe viene sempre descritta un'attività GAA inferiore a quella normale, il grado esatto di attività enzimatica residua varia fra i singoli pazienti e le diverse popolazioni di pazienti.
La Malattia di Pompe presenta un ampio spettro di fenotipi clinici ed è comunemente classificata in forma a esordio infantile (IOPD) classica e in forma a esordio tardivo (LOPD).
La IOPD classica è la forma più grave, esordisce entro i primi mesi di vita ed è caratterizzata da cardiomegalia, debolezza muscolare generalizzata e ipotonia, ed è associata a decesso, se non trattata, entro i primi due anni di vita. La LOPD per definizione può presentarsi dopo il primo anno di vita ed è caratterizzata da progressiva debolezza muscolare scheletrica ed insufficienza respiratoria5. Gungor et al,6 invece, suggeriscono di considerare la Malattia di Pompe come caratterizzata da uno spettro continuo di fenotipi: quelli a esordio precoce (a sinistra dello schema) sono più gravi e a progressione più rapida. Quelli a esordio più tardivo (a destra dello schema) sono meno gravi e a progressione più lenta. Nel fenotipo classico infantile l’esordio dei sintomi è entro il primo anno di vita, si ha sempre presenza di cardiomiopatia ipertrofica e l’attività della GAA è praticamente assente. Nel fenotipo infantile (non classico), l’esordio dei sintomi va dalla nascita all’adolescenza, ma non si osserva ipertrofia cardiaca persistente e progressiva. Nel fenotipo adulto l’esordio dei sintomi va dall’adolescenza alla tarda età.
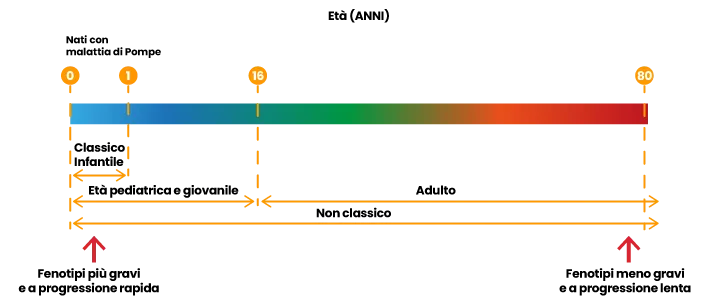
Rif. 6
I neonati con la forma infantile classica della Malattia di Pompe hanno un'attività enzimatica rilevabile minima o assente (<1%).7
I bambini e adulti con la forma ad esordio tardivo della Malattia di Pompe possono evidenziare un'attività GAA residua dell'1−30% rispetto ai livelli normali medi.7
Leggi anche:
Bibliografia
- Hirschhorn R et al. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G., eds. The Online Metabolic and Molecular Bases of Inherited Disease. OMMBID.
- Pittis MG et al. Acta Myol. 2007;26(1):67-71.
- Muller-Felber W, et al. Neuromuscul Disord. 2007,17;698-706.
- Thurberg BL et al. Lab Invest. 2006;86(12):1208-20.
- AANEM Muscle Nerve 2009; 40: 149-160.
- Gungor D et al. Am J Med Genet A 2013; 161A (2): 399-400.
- van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372(9646):1342-53.
Codice deposito aziendale MAT-IT-2100349