- Article
- Source : Campus Sanofi
- 24 mars 2024
L'ASMD : une maladie de surcharge lysosomale
L'ASMD est une maladie de surcharge lysosomale, et plus précisément une sphingolipidose, majoritairement diagnostiquée à l'âge pédiatrique et qui touche aussi bien les hommes que les femmes. Elle est décrite pour la première fois par le Dr. Albert Niemann en 1914.
C'est une maladie à transmission autosomique récessive, elle est plus fréquemment décrite dans certaines populations : Afrique du Nord et dans certains pays de l’est de l’Europe, on parle de maladie pan-ethnique.
L'ASMD est liée à une mutation biallélique du gène SMPD1, codant pour la sphingomyélinase acide, entrainant une accumulation de sphingomyéline dans les lysosomes des cellules (principalement au niveau des macrophages) dans différents organes : le foie, la rate, les poumons, la moelle osseuse et le système nerveux dans certains sous-types.
Il existe un continuum phénotypique permettant de distinguer 3 formes principales :
- L’ASMD chronique viscéral (ou ASMD de type B), représentant environ 80% des diagnostics réalisés en France ;
- L’ASMD infantile neuro-viscéral (ou ASMD de type A) représentant 12% des diagnostics en France ;
- L’ASMD chronique neuro-viscéral (ou ASMD de type A/B), forme intermédiaire représentant 8% des diagnostics en France.
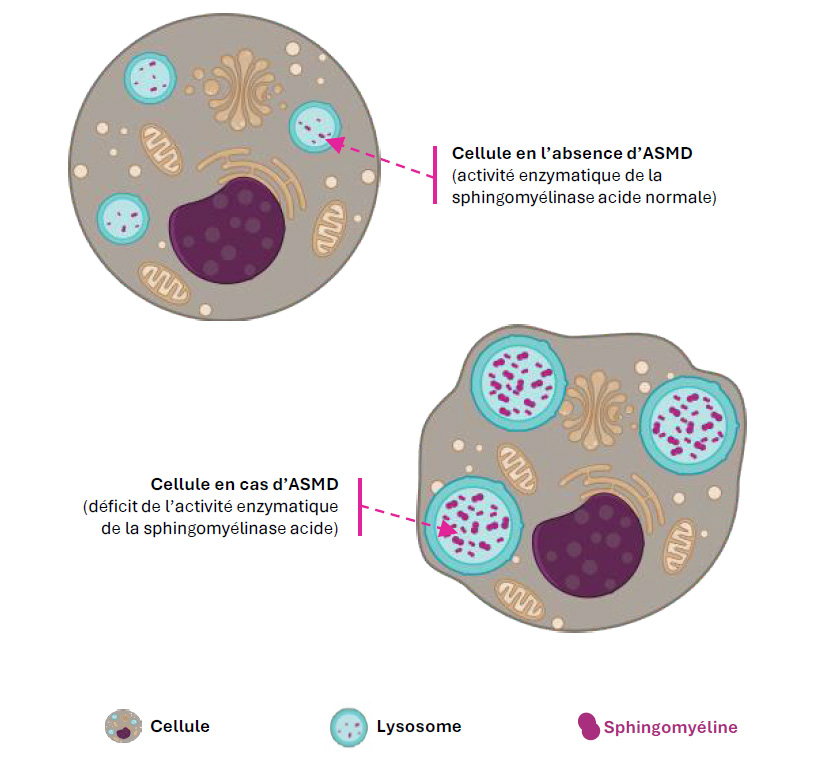
Figure 1 : Schéma simplifié du processus de surcharge lysosomale dans l'ASMD
| ASMD de type A | ASMD de type A/B | ASMD de type B | |
| Forme | Neuroviscérale infantile | Neuroviscérale chronique | Viscérale chronique |
| Phénotype | Atteinte viscérale sévère et neurologique | Atteinte viscérale progressive et neurologique variable | Atteinte viscérale progressive et sans ou peu d’atteinte neurologique |
| Apparition | Débute dans les premiers mois de vie | Débute dans l’enfance | Débute de l’enfance à l’âge adulte |
| Espérance de vie | < 3 ans | Petite enfance à l'âge adulte | Petite enfance à l’âge adulte |
En France, dans l’ASMD de type B, l’âge médian au décès est de 58,5 ans.(1)
Les patients atteints d'ASMD peuvent connaître des retards de diagnostic de 5 ans en moyenne.⁽⁶⁾
Principaux signes d'appel des formes chroniques de l'ASMD (type B et A/B) (1,2)
Les principaux signes d'appel des patients atteints d'ASMD de types B et A/B qui peuvent orienter le diagnostic sont :

> 90% des patients
Une splénomégalie
Accompagnée de saignements, d’une diminution des facteurs de coagulation et/ou à une thrombopénie.

> 90% des patients
Une Pneumopathie Interstitielle Diffuse (PID) accompagnée :
- d'opacités en verre dépoli (100% des cas)
- d'épaississements des septa interlobulaires (100% des cas)
- d'épaississements des lignes interlobulaires (90% des cas)
- de "crazy paving patterns" (40% des cas)

> 70% des patients
Une hépatomégalie
Associée à une altération progressive de la fonction hépatique.

> 50% des patients
Une thrombopénie
Associée à des saignements (dont épistaxis) et des ecchymoses.

> 70% des patients
Un profil lipidique athérogène
Associé à une activité cardiaque altérée

> 90% des patients pédiatriques
Un retard de croissance/pubertaire
> 90% des patients
Une splénomégalie
Accompagnée de saignements, d’une diminution des facteurs de coagulation et/ou à une thrombopénie.
> 90% des patients
Une Pneumopathie Interstitielle Diffuse (PID) accompagnée :
- d'opacités en verre dépoli (100% des cas)
- d'épaississements des septa interlobulaires (100% des cas)
- d'épaississements des lignes interlobulaires (90% des cas)
- de "crazy paving patterns" (40% des cas)
> 70% des patients
Une hépatomégalie
Associée à une altération progressive de la fonction hépatique.
> 50% des patients
Une thrombopénie
Associée à des saignements (dont épistaxis) et des ecchymoses.
> 70% des patients
Un profil lipidique athérogène
Associé à une activité cardiaque altérée
> 90% des patients pédiatriques
Un retard de croissance/pubertaire
Dans l'ASMD de type A/B, on retrouve des manifestations neurologiques telles que des troubles cognitifs, une hypotonie et une hyporéflexie, ainsi qu'une tâche maculaire rouge-cerise dans 1/4 des cas.
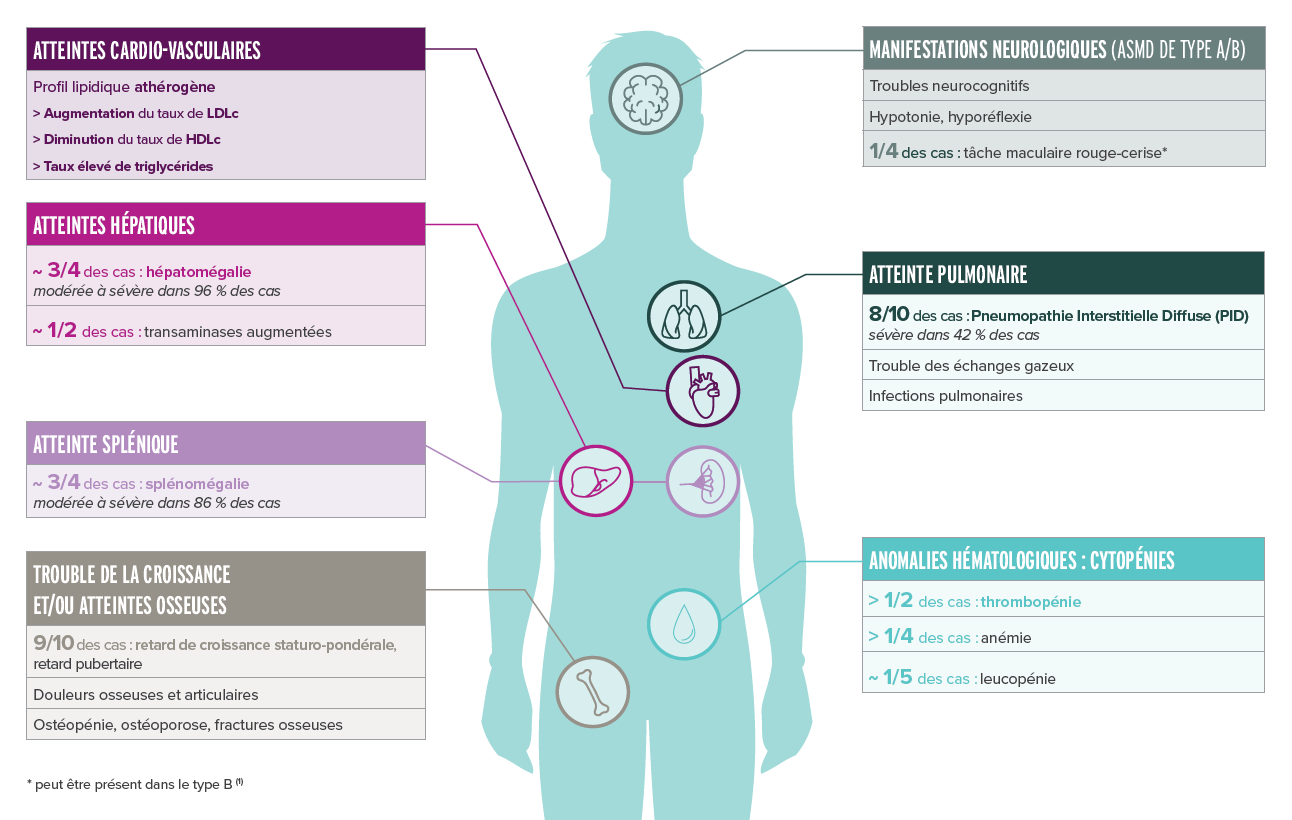
On retrouve des variabilités inter-individuelles, selon la forme de la maladie et les manifestations cliniques, ainsi que des atteintes multi systémiques fréquentes.(2,4)

Atteintes pulmonaires
28% des décès

Atteintes hépatiques
28% des décès

Complications hémorragiques
10% des décès

Autres causes
Insuffisance cardiaque, maladie neurodégénérative, cancer, complications liées à une transplantation, défaillance multiviscérale ou autres.
Atteintes pulmonaires
28% des décès
Atteintes hépatiques
28% des décès
Complications hémorragiques
10% des décès
Autres causes
Insuffisance cardiaque, maladie neurodégénérative, cancer, complications liées à une transplantation, défaillance multiviscérale ou autres.
Le risque de décès des patients atteints d'ASMD de type A/B et B est 5,5 fois plus élevé que dans la population générale.⁽⁸⁾
Un impact sur la qualité de vie des patients(9)
L’ASMD impacte également la vie des patients au niveau :
- De la fonction physique (79% des patients)
- De l’estime de soi (62%)
- Des émotions, causant anxiété, dépression, tristesse, frustration, inquiétude et peur (55%)
- De la fonction et des relations sociales (55%)
- Des soins personnels, tels que l’hygiène personnelle et la toilette, la capacité à s’habiller soi-même et à maintenir un régime alimentaire sain (31%)

ASMD : Comment diagnostiquer ?(1)
Il est recommandé de dépister l'ASMD et la maladie de Gaucher en parallèle.
Réalisé dans des laboratoires spécialisés, le diagnostic de certitude est simple et rapide.

Le saviez-vous ?
En moyenne, 1 patient sur 4 suspectés d'être atteints de la maladie de Gaucher est finalement diagnostiqué ASMD.(10)
À retenir
- L’ASMD (Déficit en sphingomyélinase acide), anciennement appelé maladie de Niemann-Pick A, A/B et B, est une maladie de surcharge lysosomale avec une incidence en France estimée à 1/200 000 naissances (tous types confondus)
- Il existe 3 formes cliniques suivant un continuum phénotypique : type A (forme neuroviscérale infantile), type A/B (forme neuroviscérale chronique) et type B (forme viscérale chronique)
- Les principaux symptômes qui peuvent orienter le diagnostic du type B sont : la splénomégalie, la Pneumopathie Interstitielle Diffuse (PID), l’hépatomégalie, la thrombopénie, les dyslipidémies athérogènes et le retard de croissance/retard pubertaire
Références
- Protocole National de Diagnostic et de Soins sur l’ASMD. Haute Autorité de Santé. Novembre 2024.
- McGovern MM, et al. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet Journal of Rare Diseases. 2017; 12:41.
- Lidove, et al. Déficit en sphingomyélinase acide (maladie de Niemann-Pick B) : une étude rétrospective multicentrique de 28 patients adultes. La Revue de Médecine Interne. 2017; 38(5):291-299.
- McGovern MM, et al. Prospective study of the natural history of chronic acid sphingomyelinase deficiency in children and adules: eleven years of observation. OrphanetJ Rare Dis. 2021: 16:212.
- McGovern MM, et al. Morbidity and mortality in type B Niemann-Pick disease. Genetics in Medicine. 2013; 15(8):618-623.
- McGovern MM, Wasserstein MP, Giugliani R, et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics. 2008;122:e341-e349.
- Simonaro C, et al. The Demographics and Distribution of Type B Niemann-Pick Disease: Novel Mutations Lead to New Genotype/Phenotype Correlations. Am. J. Hum. Genet. 2002, 71(6):1413-1419.
- Mauhin, et al. Acid sphingomyelinase deficiency in France: a retrospective survival study. Orphanet Journal of Rare Diseases. 2024; 19:289.
- Pokrzywinski R, et al. Impact and burden of acid sphingomyelinase deficiency from a patient and caregiver perspective. Sci Rep. 2021;11(1):20972.
- Oliva et al. Importance to include differential diagnostics for acid sphingomyelinase deficiency (ASMD) in patients suspected to have to Gaucher disease. Mol Genet Metab. 2023;139(1):107563
- D. Cassiman et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol Genet Metab. 2016. 118(3):206-213.
250214152842ZC - 03/2025