- Article
- Source : Campus Sanofi
- 28 févr. 2023
Diagnostiquer la maladie de Gaucher et l’ASMD*

La maladie de Gaucher et l'ASMD* (également appelée maladie de Niemann-Pick A, A/B et B.) sont deux maladies de surcharge lysosomale dues à un déficit enzymatique, entrainant une accumulation du substrat dans les macrophages. Ces macrophages surchargés en lipides s'accumulent essentiellement dans la moelle osseuse, la rate et le foie (et également les poumons dans l'ASMD). Ils conduisent à des atteintes multi-systémiques avec des manifestations viscérales, hématologiques, osseuses et pulmonaires.1-5.
* ASMD : Acid Sphingomyelinase Deficiency
Deux maladies génétiques rares du métabolisme1,2,3,4
Transmission autosomique récessive, liée à des variants pathogéniques
Sous-diagnostiquées en raison de leur rareté :
• Prévalence de 1/130 000 pour la maladie de Gaucher
• Prévalence de 1/200 000 pour l'ASMD
Hétérogénéité des manifestations
Signes d'appels communs1,4
Plusieurs signes d’appels sont communs à la maladie de Gaucher et à l’ASMD. Ces manifestations cliniques sont similaires chez l’adulte et l’enfant, à l’exception du retard de croissance ou pubertaire, observé uniquement chez l’enfant. On retrouve notamment :
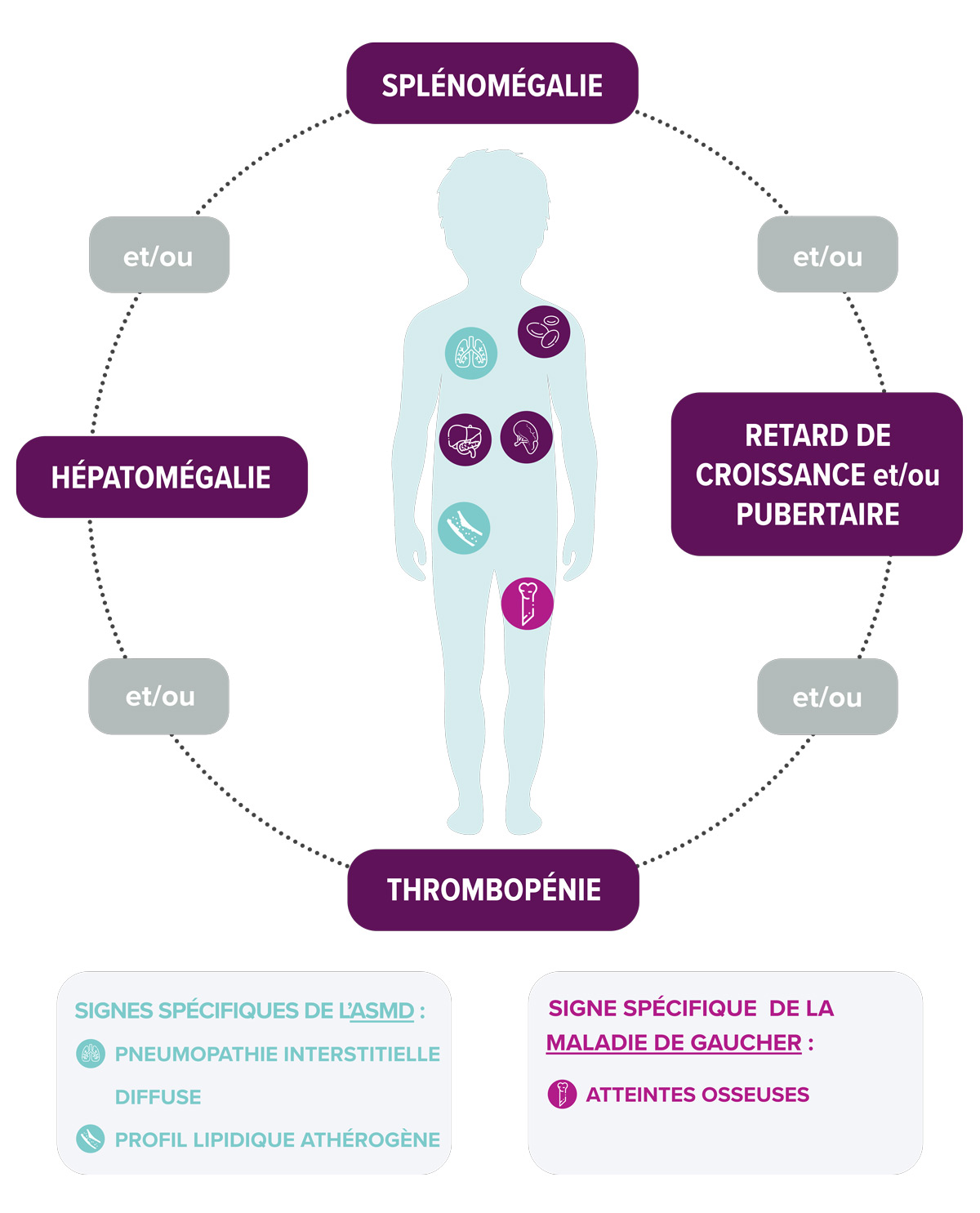
L’absence d’un ou plusieurs de ces signes ne doit pas exclure le diagnostic.
Selon les recommandations des PNDS de la maladie de Gaucher et de l’ASMD :
En cas de suspicion et en raison d’une présentation clinique pouvant être très proche, le dosage de l’activité des deux enzymes est recommandé.
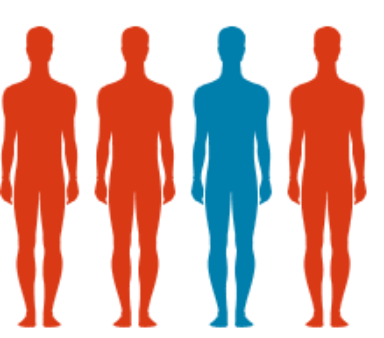
En moyenne, 1 patient
sur 4 suspectés d’être atteint de la maladie de Gaucher
est finalement diagnostiqué ASMD. (11)
Pour en savoir plus sur le diagnostic de ces deux pathologies veuillez consulter les documents suivant :
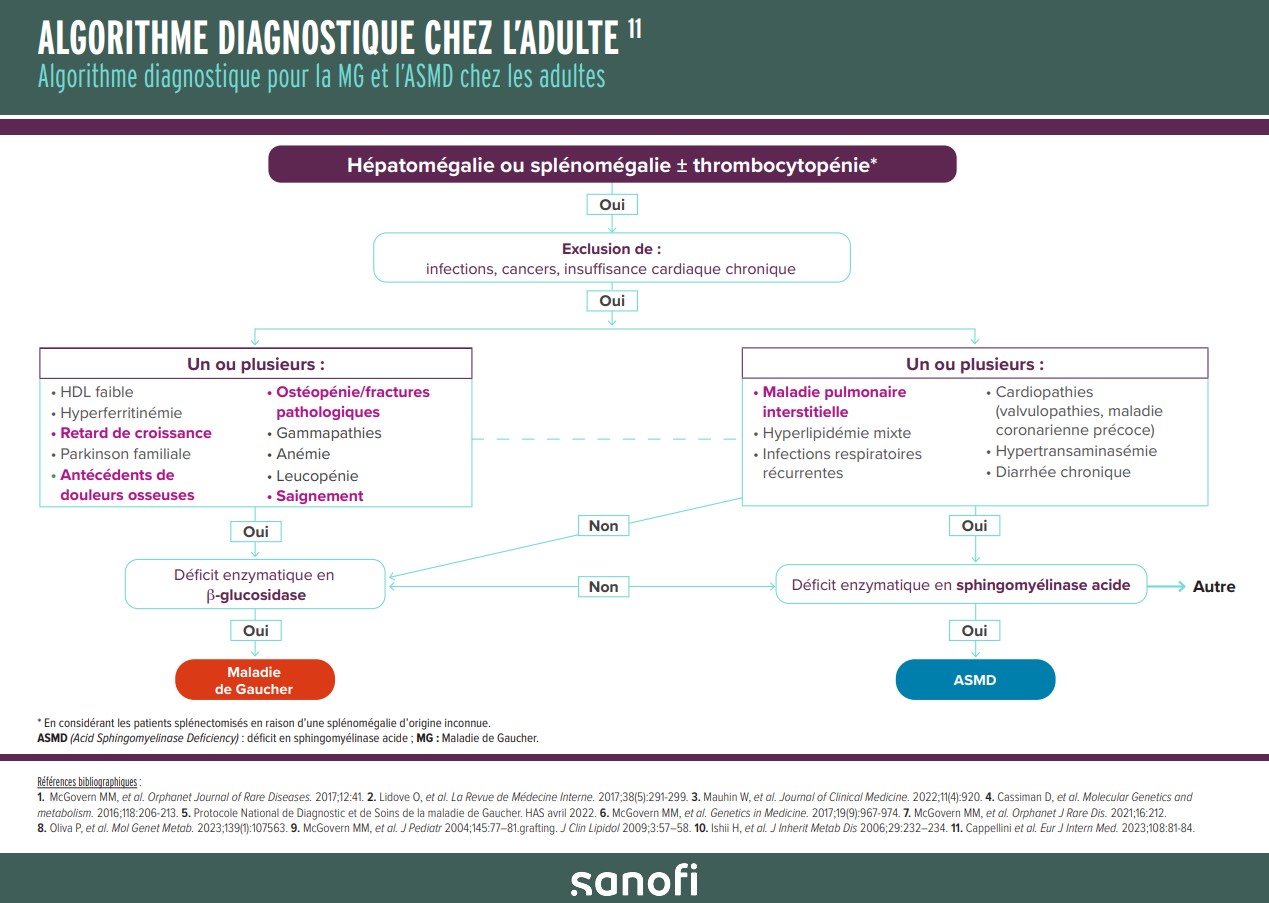
Algorithmes diagnostiques adulte et pédiatriques
Téléchargez ces deux fiches d’intérêts concernant l’ASMD* : définition, présentation clinique, symptômes orientant le diagnostic et diagnostic de certitude ; ainsi que les algorithmes diagnostiques adulte et pédiatrique, mettant en avant l’importance du parallel testing entre la Maladie de Gaucher et l’ASMD
Comment confirmer le diagnostic1,2 ?
Le diagnostic de certitude repose sur la mesure de l’activité des enzymes spécifiques dans la maladie de Gaucher et l’ASMD.
Maladie de Gaucher
Mesure de l'activité enzymatique de la glucocérébrosidase
(ou Beta-glucosidase acide)
ASMD
Mesure de l'activité enzymatique de la sphingomyélinase acide
Comment réaliser les dosages enzymatiques1 ?
Ils peuvent être effectués sur tube EDTA ou sur papier buvard
Un diagnostic précoce est essentiel pour optimiser la prise en charge de ces maladies⁴,⁸⁻⁹.
Des anomalies cytologiques peuvent orienter le diagnostic1,2
On peut retrouver au myélogramme :
- Dans la Maladie de Gaucher : des cellules de Gaucher
- Dans l'ASMD : des histiocytes “bleu de mer” et des macrophages spumeux.
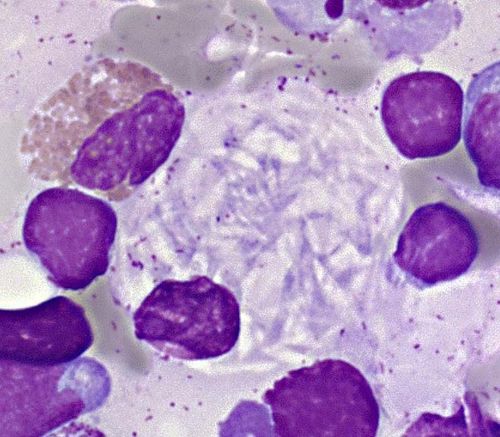
Cellule de Gaucher : Myélogramme d'un patient atteint de la maladie de Gaucher
Avec l'aimable autorisation du Dr Munier - Biologiste - CH Valence
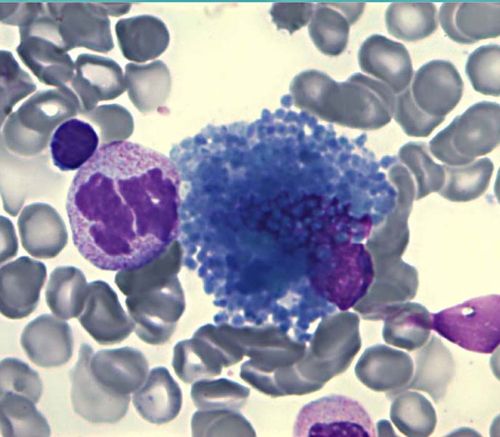
Histiocyte bleu de mer : Myélogramme d'un patient ASMD type B
Avec l'aimable autorisation du Dr Lours - Biologiste en hématologie Cellulaire - CH Lyon Sud, HCL
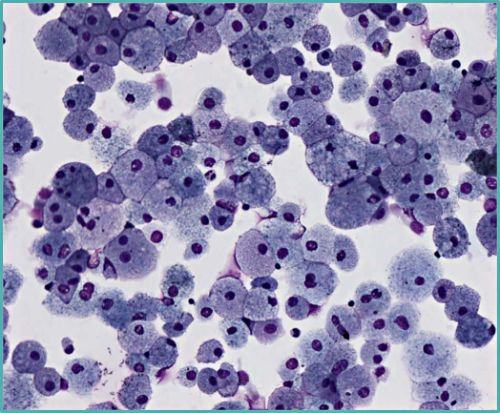
Macrophages spumeux : LBA d'une patiente ASMD de type B
Avec l’aimable autorisation du Dr Guyard – Anatomopathologiste à l’hôpital Bichat-Claude Bernard, AP-HP
Dans la recherche de l'ASMD : une hypercellularité et des macrophages spumeux peuvent être retrouvés au lavage broncho-alvéolaire (LBA).

Un myélogramme ou un LBA ne permet pas de confirmer le diagnostic de la maladie de Gaucher ou de l’ASMD.
Seule la mesure de l'activité enzymatique permet de poser le diagnostic de certitude1,2.

Références
- Protocole National de Diagnostic et de Soins de la maladie de Gaucher. HAS avril 2022
- Protocole National de Diagnostic et de Soins de l’ASMD. HAS novembre 2024.
- Lidove et al. Déficit en sphingomyelinase acide (Maladie de Niemann-Pick B) : une étude retrospective multicentrique de 28 patients adultes. La Revue de Médecine Interne. 2017;38(5):291-299.
- McGovern et al. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genetics in Medicine. 2017;19(9);967-974.
- McGovern et al. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet Journal of Rare Diseases. 2017;12(1):41
- Cappellini MD et al. Finding and Treating Gaucher Disease Type 1 - The Role of the Haematologist. European Oncology & Haematology 2018;14:50-56
- McGovern MM et al. A Prospective, Cross-sectional Survey study of the natural history of Niemann-Pick Disease Type B. Pediatrics. 2008;122(2):e341-e349
- Gaucher Registry Annual Report 2009
- Mistry et al. Disease state awareness in Gaucher disease: A Q&A expert roundtable discussion. Clinical Advances in Hematology & Oncology. 2012;10(6 Suppl 8):1-16.
- McGovern et al. Morbidity and mortality in type B Niemann–Pick disease. Genetics in Medicin. 2013;15(8):618-623.
- Oliva et al. Importance to include differential diagnostics for acid sphingomyelinase deficiency (ASMD) in patients suspected to have to Gaucher disease. Mol Genet Metab. 2023;139(1):107563
250424161015UN - 06/2025