- Article
- Source : Campus Sanofi
- 3 juil. 2025
La maladie de Gaucher : une maladie de surcharge lysosomale
Qu'est ce que la maladie de Gaucher1-3 ?
La pathologie a été décrite pour la première fois par le Dr Philippe GAUCHER en 18822. C'est une maladie de surcharge lysosomale causée par un déficit en glucocérébrosidase (ou glucosylcéramidase ou β-glucosidase acide, GBA) ou exceptionnellement en son activateur, la saposine C.
La glucocérébrosidase permet d’hydrolyser le glucosylcéramide (ou glucocérébroside), sphingolipide issu de la dégradation des membranes cellulaires, en céramide (ou cérébroside) et glucose. Dans la maladie de Gaucher, le glucosylcéramide non dégradé s’accumule principalement dans les lysosomes des macrophages. Ces derniers adoptent alors une morphologie caractéristique (cellules de Gaucher)1.

Cette accumulation du substrat dans les macrophages entraîne une atteinte multisystémique3 (foie, rate, moelle osseuse...).
Les différents phénotypes 1,4
L’expression clinique est variable et le diagnostic peut être évoqué à tout âge. On distingue trois principaux phénotypes :
- Le type 1 : son expression clinique est très hétérogène allant de formes asymptomatiques tout au long de la vie à des formes sévères dès l’enfance. Il peut associer à des degrés variables une organomégalie, des cytopénies et des atteintes osseuses.
- Le type 2 : il s’agit d’une forme exceptionnelle, d’expression très précoce et de mauvais pronostic.
- Le type 3 : c’est une forme rare associant une encéphalopathie progressive de sévérité variable et des manifestations communes au type 1.
| Type 1 | Type 2 | Type 3 | |
| Âge d'apparition des symptômes | Enfance-Adulte | < 1 an | Enfance-Adolescence |
| Atteinte neurologique | Non | Sévère | Modérée à sévère |
| Espérance de vie | 60 ans | < 3 ans | < 20 – 30 ans |
| Fréquence | 95% | < 1% | 5% |
Adapté de Stirnemann et al., Aspects épidémiologiques, cliniques, biologiques et thérapeutiques de la maladie de Gaucher. 2003;32:503-11, et du Protocole National de Diagnostic et de soins de la maladie de Gaucher. Avril 2022.
La maladie de Gaucher : symptômes chez l’enfant et l'adulte 1
Les principaux signes d’appel des patients atteints de la maladie de Gaucher sont les suivants :
- Une organomégalie : augmentation des volumes du foie et de la rate ;
- Des cytopénies : principalement des thrombopénies5 et des anémies ;
- Des atteintes osseuses à l'origine de douleurs aiguës sous forme de crises osseuses et/ou de douleurs chroniques osseuses et articulaires. (Une ostéopénie ou une ostéoporose peuvent survenir chez des patients jeunes.)
Chez l'enfant1,6
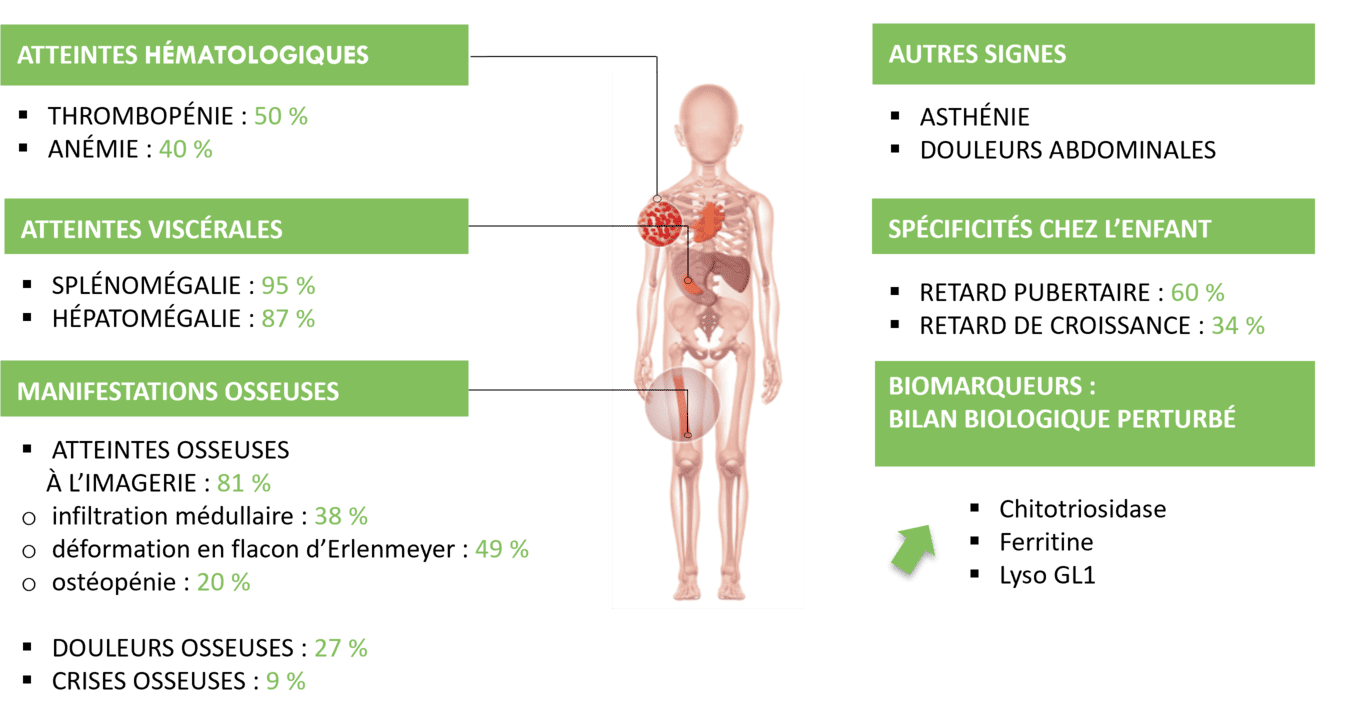
Adapté de Stirnemann et al., Aspects épidémiologiques, cliniques, biologiques et thérapeutiques de la maladie de Gaucher. 2003;32:503-11, et du Protocole National de Diagnostic et de soins de la maladie de Gaucher. Avril 2022.
* L’absence d’un ou plusieurs de ces signes n’exclut pas le diagnostic.
D’autres atteintes sont à considérer (liste non exhaustive) : autres atteintes hématologiques, asthénie, hyperferritinémie, autres atteintes osseuses à l’imagerie (ostéoporose…), atteintes neurologiques dans la maladie de Gaucher de type 3 (apraxie oculomotrice, épilepsie, ataxie, ophtalmoplégie, spasticité, démence…)
Chez l'adulte1,7-8
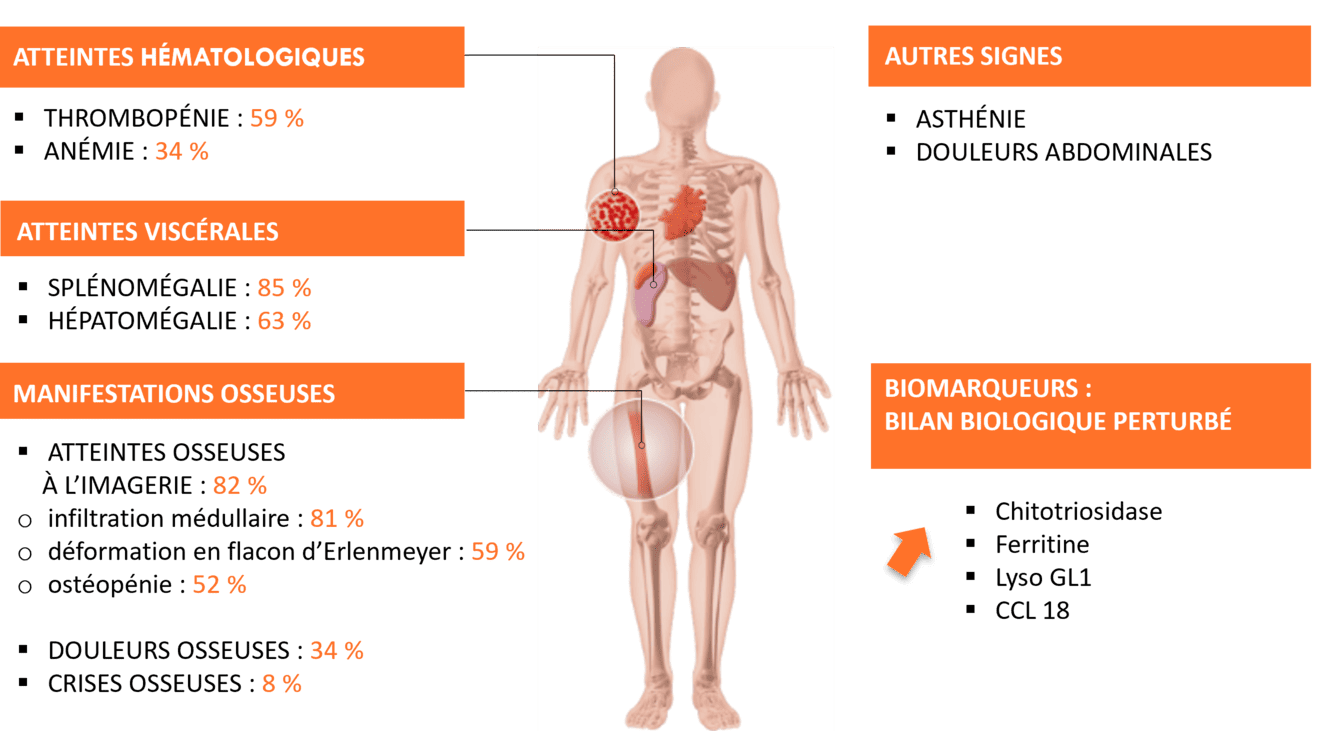
Adapté du rapport annuel du registre de Gaucher de 2009
*L'absence d'un ou plusieurs de ces signes n'exclut pas le diagnostic.
D'autres atteintes sont à considérer (liste non exhaustive) : autres atteintes hématologiques (gammapathies monoclonales à signification indéterminée), hyperferritinémie, syndrome parkinsonien, antécédent de la maladie de parkinson.
Chaque patient est unique en termes d'âges d'apparition et de sévérité des symptômes9.

22 ans 1,10
C’est l’âge médian au moment du diagnostic.*

Jusqu’à 10 ans 11
C’est la durée que peut atteindre l’errance diagnostique des patients.

A tout âge
Le diagnostic peut être réalisé, que ce soit chez l’enfant ou l’adulte.
* diagnostic effectué entre 0 et 84 ans
Atteintes osseuses et articulaires : complications majeures de la maladie de Gaucher 7,9
Non diagnostiquée, la maladie de Gaucher peut entraîner des complications osseuses irréversibles11.
L’atteinte ostéoarticulaire est une complication majeure de la maladie de Gaucher de type 1 :
- Altération de la qualité de vie
- Cause de morbidité
- Invalidité, douleurs
- Limitations articulaires
- Hospitalisation
Analyse de 602 patients du registre international de la maladie de Gaucher9

UN DIAGNOSTIC PRÉCOCE EST ESSENTIEL POUR PRÉVENIR L’APPARITION DE COMPLICATIONS OSSEUSES IRRÉVERSIBLES11
Maladie de Gaucher : comment diagnostiquer ?1
Le diagnostic biologique repose sur la mise en évidence d’un déficit de l’activité de la glucocérébrosidase dans le sang et est réalisé à tout âge dans des laboratoires spécialisés.
Comment ?
- Sur tâches de sang séché (buvard)
- Sur tube de sang (EDTA)

Le saviez-vous ?
En moyenne, 1 patient sur 4 suspectés d’être atteint de la maladie de Gaucher est finalement diagnostiqué ASMD. 12
Il est recommandé de dépister l'ASMD et la maladie de Gaucher en parallèle.12
Réalisé dans des laboratoires spécialisés, le diagnostic de certitude est simple et rapide.
À retenir
- La maladie de Gaucher est une maladie de surcharge lysosomale à transmission autosomique récessive. Son incidence annuelle moyenne est de l'ordre de 1/50 000 naissances dans la population générale.
- On distingue trois principaux phénotypes : les types 1, 2 et 3.
- Les principales atteintes liées à la maladie de Gaucher sont les manifestations osseuses, l'hépatosplénomégalie ainsi que la thrombopénie.
Références
1. Protocole National de Diagnostic et de Soins de la maladie de Gaucher. Avril 2022.
2. Gaucher, Philippe-Charles-Ernest. De l’épithélioma primitive de la rate : Hypertrophie idiopathique de la rate sans leucémie. Thèse de doctorat en médecine. Paris : Faculté de médecine, 1882, 31p.
3. Mistry et al. Disease state awareness in Gaucher disease: A Q&A expert roundtable discussion. Clinical Advances in Hematology & Oncology 2012;10(6).
4. Stirnemann et al. Aspects épidémiologiques, cliniques, biologiques et thérapeutiques de la maladie de Gaucher. La Presse Médicale. 2003;32 :503- 511.
5. Belmatoug et al. La maladie de Gaucher. Journal de la Société de Biologie. 2002 ;196(2) :141-149
6. Kaplan P, et al. The Clinical and Demographic Characteristics of Non neuronopathic Gaucher Disease in 887 Children at Diagnosis. Arch Pediatr Adolesc Med. 2006;160:603-608
7. Gaucher Registry Annual Report 2009.
8. Stirnemann. Nouveauté sur la maladie de Gaucher. Revue de médecine interne. Rev Med Interne. 2008;29(3):176-8.
9. Javier et al. Atteintes ostéoarticulaires de la maladie de Gaucher chez l’adulte : de la physiopathologie au traitement. Presse Med. 2007;36:1971-1984.
10. Stirnemann et al. The French Gaucher’s Disease registry: clinical characteristics, complications and treatment of 562 patients. Orphanet Journal of Rare Disease 2012;7:77
11. Mistry et al. Consequences of diagnostic delays in type 1 Gaucher disease: The need for greater awareness among Hematologists-Oncologists Am. J. Hematol. 2007;82:697-701
12. Oliva et al. Importance to include differential diagnostics for acid sphingomyelinase deficiency (ASMD) in patients suspected to have to Gaucher disease. Mol Genet Metab. 2023;139(1):107563
250411172236DT - 05/2025