- Artículo
- Fuente: Campus Sanofi
- 21 may 2024
Reto clínico en ASMD

Autor:
Dr. Miguel García González
Introducción
Paciente mujer de 33 años con hepatoesplenomegalia diagnosticada a los 16 años de origen desconocido. Consulta por dolor abdominal y dificultad respiratoria con el ejercicio. En la analítica destaca bilrrubina total 2,3 mg/dl AST 51 U/l, ALT 60 U/l, GGT 10/U/L, LDH 204 U/I, plaquetas 128.000 / MicroL, Colesterol 350 mg/dl , HDL 22 mg/dl, LDL 328 mg/dl, TG 390 mg/dl. El fibroscan mostró valores de 16 kPa y CAP de 340 compatibles con Fibrosis 4 y esteatosis grado 3. En la espirometría muestra un FVC de 70% con FEV1/FVC 90% compatible con patrón restrictivo y un DLco disminuido. En TAC body se objetiva arterioesclerosis generalizada . La densitometría ósea mostró osteoporosis.
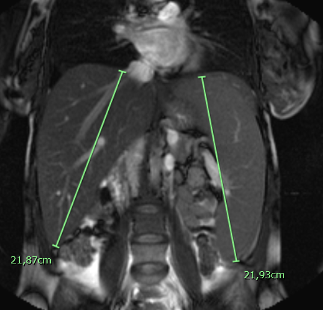
Señale cuál es el diagnóstico correcto:
Respuesta incorrecta. Sigue intentándolo
Respuesta incorrecta. Sigue intentándolo.
Respuesta incorrecta. Sigue intentándolo.
Respuesta correcta: Déficit de esfingomielinasa ácida
El diagnóstico diferencial se establece fundamentalmente entre las dos enfermedades de depósito. El resto de las opciones, por la larga evolución y tipo de afectación pulmonar y hepatoesplénica se descartan. Existen signos y síntomas comunes a ambas enfermedades de depósito lisosomal, Gaucher y ASMD, como son la hepatoesplenomegalia, trombopenia y osteoporosis. Pero la afectación pulmonar restrictiva ascociada a enfermedad pulmonar intersticial, la enfermedad hepática potencialmente progresiva, la arterioseclerosis y la dislipemia con niveles de HDL anormalmente bajos son típicos de ASMD (déficit de esfingomielinasa ácida por mutaciones en el gen SMPD1) y en la enfermedad de Gaucher el curso de la afectación pulmonar es leve.
En ASMD los datos de gravedad son el empeoramiento de la disnea, las plaquetas <50.000, la hemorragia recurrente, el daño biológico o histológico hepático, y la esplenomegalia dolorosa.
Respueta incorrecta. Sigue intentándolo
Autor

Dr. Miguel García González
Médico Adjunto del Servicio de Aparato Digestivo
Hospital Universitario Ramón y Cajal. Madrid

Referencias
1. Kingma, S.D.; Bodamer, O.A.; Wijburg, F.A. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 145–157.
2. Margaret M. McGovern, Carlo Dionisi-Vici, Roberto Giugliani, et als. Consensus recommendation for a diagnostic guideline foracid sphingomyelinase deficiency. Genetics in Medicine 2017: 19: 967-974
3. Matthew D. Jankowich, Barry S. Shea, Interstitial Lung Diseases. Ed. By Misra, Abhinav Kumar; Jankowich, Matthew D.; Shea, Barry S. In Cecil Essentials of Medicine 10 Ed. Philadelphia. Elsevier 2022. Capítulo 17, 199-215
4. Robert J. Hopkin; Gregory A. Grabowski. Enfermedades por almacenamiento lisosómico. Ed by Joseph Loscalzo, Anthony Fauci, Dennis Kasper, Stephen Hauser, Dan Longo, J. Larry Jameson. . In Harrison. Principios de Medicina Interna, Ed 21., New York, NY: McGraw-Hill 2022 , Cap 418 ,
5. Tarekegn Geberhiwot , Melissa Wasserstein, Subadra Wanninayake et als. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann–Pick disease types A, B and A/B). Orphanet Journal of Rare Diseases (2023) 18:85. Disponible en https://doi.org/10.1186/s13023-023-02686-6.
MAT-ES-2401559 V1 Mayo 2024