- Artículo
- Fuente: Campus Sanofi
- 26 mar 2024
Genética y epidemiología de la MPS I

La MPS I es un trastorno autosómico recesivo
Lo que significa que hay un 25% de posibilidades de que un niño nazca con MPS I cuando ambos padres son portadores de un gen afectado. La incidencia global de la MPS I es de aproximadamente 1 por cada 100.000 recién nacidos.1
Incidencia y prevalencia
La incidencia global de la MPS I es de aproximadamente 1 por cada 100.000 recién nacidos.1 Sin embargo, los datos epidemiológicos son escasos. Aunque la enfermedad se reparte por todas las regiones geográficas y grupos étnicos, la incidencia y la distribución de los fenotipos varían de una región a otra.1-5 La incidencia más alta conocida corresponde a la población de nómadas irlandeses, con una frecuencia de portadores de 1:10 y una incidencia de la enfermedad de 1:371.5
Patrón de herencia
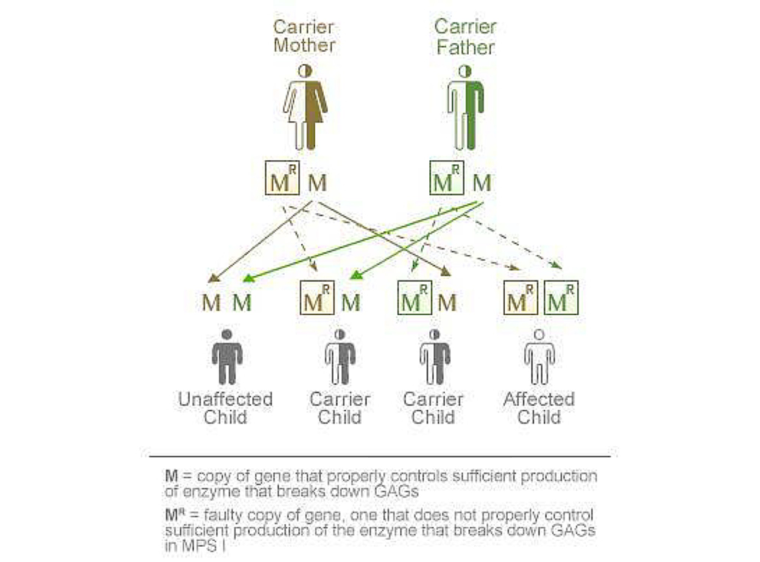
La MPS I se hereda de forma autosómica recesiva, lo que significa que la enfermedad aparece en una persona que hereda dos copias defectuosas del gen de la α-L-iduronidasa (Fig. 1).6 Ambos progenitores de un niño afectado por MPS I serán heterocigotos (portadores) para la mutación que causa la enfermedad en el gen de la α-L-iduronidasa. Tendrán un gen normal y un gen mutado, por lo que serán asintomáticos, ya que la única copia funcional del gen permite a la persona producir cantidades suficientes de la enzima α-L-iduronidasa. Cada hijo de una pareja en la que ambos padres son heterocigotos para la mutación causante de enfermedad en el gen de la α-L-iduronidasa tiene un 25% de probabilidades de resultar afectado por la MPS I, un 50% de probabilidades de ser un portador heterocigoto sin afectación y un 25% de probabilidades de no ser portador (homocigoto) ni presentar afectación. El hermano no afectado de una persona con MPS I tiene una probabilidad del 67% de ser portador heterocigoto y una probabilidad del 33% de no ser portador ni presentar afectación.6 En una misma familia, todos los hermanos afectados tendrán el mismo genotipo. No obstante, se han descrito variaciones notables de las manifestaciones de la enfermedad entre hermanos con las mismas mutaciones.

¿Tienes alguna pregunta? Escríbenos para resolver tus dudas.
Consulta de Información Médica
Referencias
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249-54.
- Moore D, et al. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis 2008;3:24.
- Lin HY, Lin SP, Chuang CK, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984-2004. Am J Med Genet A 2009;149A:960-4.
- Malm G, Lund AM, Mansson JE, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta Paediatr 2008;97:1577-81.
- Murphy AM, Lambert DM, Treacy EP, O’Meara A, Lynch SA. Incidence and prevalence of mucopolysaccharidosis type 1 in the Irish Republic. Arch Dis Child 2009;94:52-4.
- Neufeld EF and Muenzer J. (2001) The mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, and Vogelstein B. (eds.). 8th edition, Vol. III. McGraw-Hill, Medical Publishing Division, pp. 3421
MAT-ES-2503840 V1 Diciembre 2025