- Artículo
- Fuente: Campus Sanofi
- 21 mar 2024
Diagnóstico de enfermedad de Fabry

¿Quién puede diagnosticar la enfermedad de Fabry?
La enfermedad de Fabry es un trastorno multisistémico, que afecta a muchos órganos diferentes del organismo. Por lo tanto, los pacientes suelen consultar a varios especialistas antes de obtener un diagnóstico. El diagnóstico puede requerir la colaboración y la comunicación entre genetistas, asesores genéticos, nefrólogos, neurólogos, pediatras, dermatólogos, cardiólogos, internistas, oftalmólogos, gastroenterólogos, entre muchas otras. Además, debido a la naturaleza progresiva de la enfermedad de Fabry, el diagnóstico temprano es importante para permitir llevar a cabo las intervenciones adecuadas y tratar de prevenir afecciones irreversibles.
¿Cuándo sospechan los especialistas que es Fabry?
Punto de vista Neuropediatra
Dr. Daniel Natera De Benito
Neuropediatría
Hospital Sant Joan de Deu, Barcelona
Los primeros síntomas de enfermedad de Fabry aparecen a los 6 años o incluso antes. Los más característicos en niños son:
- Dolor neuropático quemante o punzante en manos y pies preferentemente
- Dolor crónico con agudizaciones recurrentes o “crisis Fabry”, espontánea o provocada por ejercicio, calor o fiebre
- Córnea verticilata
- Alteraciones motilidad intestinal: dolor abdominal inespecífica que por si sola no hace sospechar Fabry
- Hipohidrosis que no suele ser motivo de consulta
- Angioqueratomas, generalmente en zona abdomina o del bañador
- Proteinuira en algunos pacientes, pero no es lo más habitual
La mayoría de los pacientes Fabry son diagnosticados de adultos y refieren haber tenido síntomas desde niños, incluso haber consultado por ellos, siendo el dolor neuropático el síntoma más común. Es importante que los pediatras los conozcan para poder ayudar a estos pacientes lo antes posible.
Punto de vista Nefróloga
Dra. Marián Goicoechea
Nefrología
Hospital Gregorio Marañón, Madrid
La enfermedad de Fabry es una enfermedad metabólica rara y es aún más rara si no pensamos en ella. Detectarla y tratarla puede salvar una vida. Hay unos 300 diagnosticados en España pero la prevalencia según literatura es de 1 caso por cada 40.000 habitantes por lo que debería haber unos 1100 pacientes. Se trata, pues, de una enfermedad infradiagnosticada. Algunos signos deberían hacer sospechar Fabry al nefrólogo:
- Historia familiar de nefropatía
- Pacientes jóvenes con insuficiencia renal no filiada, proteinuria o albuminuria
- Isostenuria, disfunción tubular e hiperfiltración glomerular
- Microhematuria, quistes o síndrome nefrótico
- Sin hipertensión arterial generalmente
- Síntomas extranefrológicos
Diagnóstico diferencial con:
- Nefropatía IgA
- Síndrome Alport
- Glomeruesclerosis segmentaria y focal
- Lupus eritematoso sistémico
- Vasculitis
La enfermedad de Fabry tiene tratamiento, a diferencia de otras nefropatías hereditarias. Nuestra sospecha diagnóstica puede cambiar el futuro de estos pacientes.
Punto de vista Cardiólogo
Dr. Luis Tamargo
Cardiología
Hospital de la Serranía de Ronda, Málaga
La prevalencia de las afectaciones cardíacas en Fabry hace muy probable que los cardiólogos nos encontremos con algún caso a lo largo de nuestra vida profesional. Debemos conocer sus signos y síntomas cardiacos más frecuentes para evitar que nos pase desapercibido.
- Forma de presentación y evolución de la cardiopatía Fabry
- Miocardiopatía hipertrófica concéntrica aislada (sin HTA, ni comorbilidades)
- Cardiopatía isquémica con coronarias normales
- Afectación difusa: miocardio, válvulas, arritmias
- Realce tardío infero-lateral basal, intramural y subepicárdico
- PR corto y T negativas
- Síntomas extra-cardiacos
- Afectación renal
- Afectación SNC (accidente cerebrovascular y lesiones en sustancia blanca)
- Dolor neuropático y crisis febriles en la infancia
- Angioqueratomas
- Cornea verticilata
- Anamnesis e historia familiar
- Integrar antecedentes familiares: nefropatía, ictus, arritmias, muertes prematuras
- Establecer estudio inicial con familiares sintomáticos
- Correlacionar hallazgos con genética, biomarcadores y género
La enfermedad de Fabry tiene tratamiento, a diferencia de otras cardiopatías familiares. Nuestra sospecha diagnóstica puede cambiar el futuro de estos pacientes.
Punto de vista Neurólogo
Dr. Tomás Pérez Concha
Neurología
Hospital de Cruces, Vizcaya
Afectación Sistema Nervioso Periférico (SNP): neuropatía de fibra fina. Los síntomas son:
- Acroparestesias en la primera década de la vida: se suelen atribuir erróneamente a dolores de crecimiento y retrasar el diagnóstico. Hay que pensar en Fabry y buscar más síntomas
- Crisis de dolor abdominal y fiebre
- Exacerbaciones de dolor esponténamente o tras ejercicio, calor, fiebre
- Hipohidrosis o anhidrosis: reducción de sudoración, lágrimas y saliva y dificultad para regular la temperatura
- Intolerancia al ejercicio
- Crisis de diarrea o estreñimiento
Afectación del Sistema Nervioso Central (SNC)
- Ictus agudo entre 30 y 40 años, sin factores de riesgo. Generalmente, ictus isquémico pero puede ser hemorrágico. Es la primera manifestación grave de Fabry en el 70% de los casos
- Leucopatía isquémica en el 50% de los casos y asintomática, en paciente menor de 50 años
- Signo de pulvinar: hiperintensidad en secuencias T1 de la resonancia o calcificaciones en el TAC cerebral en el núcleo talámico pulvinar
- Dolicoectasia intracraneal basilar en el 20% de los pacientes
Diagnóstico diferencial1
Los síntomas pueden aparecer en la infancia y podrían no reconocerse o atribuirse a otras causas o enfermedades. El resultado puede ser que el diagnóstico se retrase y que ya existan daños en los órganos. Además, el amplio espectro de fenotipos clínicos existente y la heterogeneidad en la aparición y la progresión entre diferentes pacientes añade complejidad al diagnóstico.
El diagnóstico temprano de la enfermedad de Fabry es importante para limitar el riesgo de daño orgánico permanente, de morbilidad temprana y de muerte.
Anamnesis dirigida
Ante un signo o síntoma sospechoso de la enfermedad de Fabry, un par de preguntas2 sobre el historial clínico y antecedentes familiares de los paientes son suficientes para incrementar la sospecha del diagnóstico.
¿Alguna vez ha sufrido usted o tiene un familiar con problemas de riñón?
¿Alguna vez ha sufrido usted o tiene un familiar con problemas de corazón?
Descarga la anamnesis dirigida completa en la enfermedad de Fabry

El cribado clínico y genético de los miembros en riesgo de familias de pacientes Fabry recién diagnosticados es de crítica importancia, ya que se pueden identificar miembros adicionales de la familia con la enfermedad de Fabry a partir de cada paciente individual3.
Un estudio de revisión de pedigrí encontró que, en promedio, había al menos cinco miembros de la familia diagnosticados con la enfermedad de Fabry luego del diagnóstico de un probando3.
Sin embargo, si bien unos antecedentes familiares positivos son un sólido indicador de que se trata de la enfermedad de Fabry, se han documentado mutaciones de novo o espontáneas. Por lo tanto, la ausencia de antecedentes familiares no descarta un diagnóstico de la enfermedad de Fabry4.
Un paciente Fabry, una familia susceptible de padecer Fabry
Dr Roberto Barriales- Cardiólogo en Unidad de Cardiopatías Familiares (Complejo Hospitalario Universitario A Coruña) - destaca la importancia del estudio familiar en la Enfermedad de Fabry.
Liso-GL-3, biomarcador de la Enfermedad de Fabry
La enfermedad de Fabry provoca la acumulación de GL-3 predominantemente en los lisosomas de varios tipos de células4.
A día de hoy, el liso-GL-3 es un biomarcador más sensible, que puede correlacionarse con la gravedad de la enfermedad o la afectación orgánica en la enfermedad de Fabry. El liso-GL-3, también conocido como globotriaosilesfingosina, es una forma deacilada de GL-35.
Pruebas diagnósticas DBS
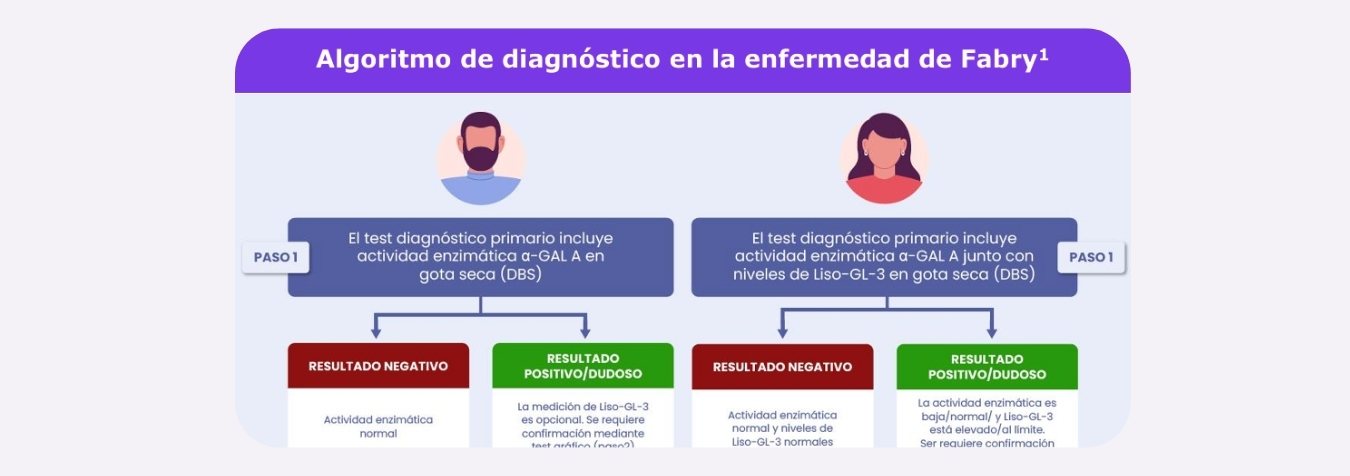
Descarga a continuación la infografía algoritmo de diagnóstico:
Adaptación de Balendran S, et al. Clin Genet. 2020
* Grupos especiales de pacientes pueden requerir un seguimiento de pruebas genéticas
independientemente de los valores bioquímicos.

Referencias
- Thomas AS. Difficulties and barriers in diagnosing Fabry disease: what can be learnt from the literature? Expert Opin Med Diagn 2013;7(6):589-99.
- Michaud M, et al. When and How to Diagnose Fabry Disease in Clinical Pratice. Am J Med Sci. 2020
- Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018
- Germain DP. Fabry Disease. Orphanet J Rare Dis. 2010 Nov;5(1):30.
- Rombach SM, et al. plasma globotriaosylsphyngosine: diagnostic value and relation to clinical manifestation in fabry. Biochim Biophys Acta 2010; 1802(9):741-8
- Germain DP. Orphanet J Rare Dis 2010; 5: 30.
- Nowak A et al. Mol Genet Metab 2017; 120 (1-2): 57-61.
- Laney DA et al. J Genet Couns 2013; 22(5): 555-64.
- Deegan PB et al. J Med Genet 2006; 43(4): 347-352.
- Linhorst GE et al. Clin Chim acta 2005: 353(1-2): 201-203.
MAT-ES-2303247 V1 Febrero 2024