
Indicación de Fabrazyme®
El primer tratamiento en demostrar una tasa sustancialmente menor de acontecimientos renales, cardíacos, cerebrovasculares y muertes en la población por protocolo frente a la tratada con placebo, en pacientes con enfermedad de Fabry.1-3
Fabrazyme está indicado como terapia de sustitución enzimática a largo plazo en pacientes con diagnóstico confirmado de enfermedad de Fabry (déficit de a-galactosidasa A).
Fabrazyme está indicado en adultos, adolescentes y niños de 8 años o mayores.
Estudios sobre Fabrazyme®
.png)
Reducción de eventos graves
Reducción del 61% del riesgo relativo de acontecimientos clínicos renales, cardíacos y cerebrovasculares potencialmente mortales y muerte en pacientes tratados con Fabrazyme® pacientes frente a placebo*, 2.
Un porcentaje menor de pacientes tratados con Fabrazyme® (27%) experimentó eventos clínicos en comparación con placebo (42%)2.
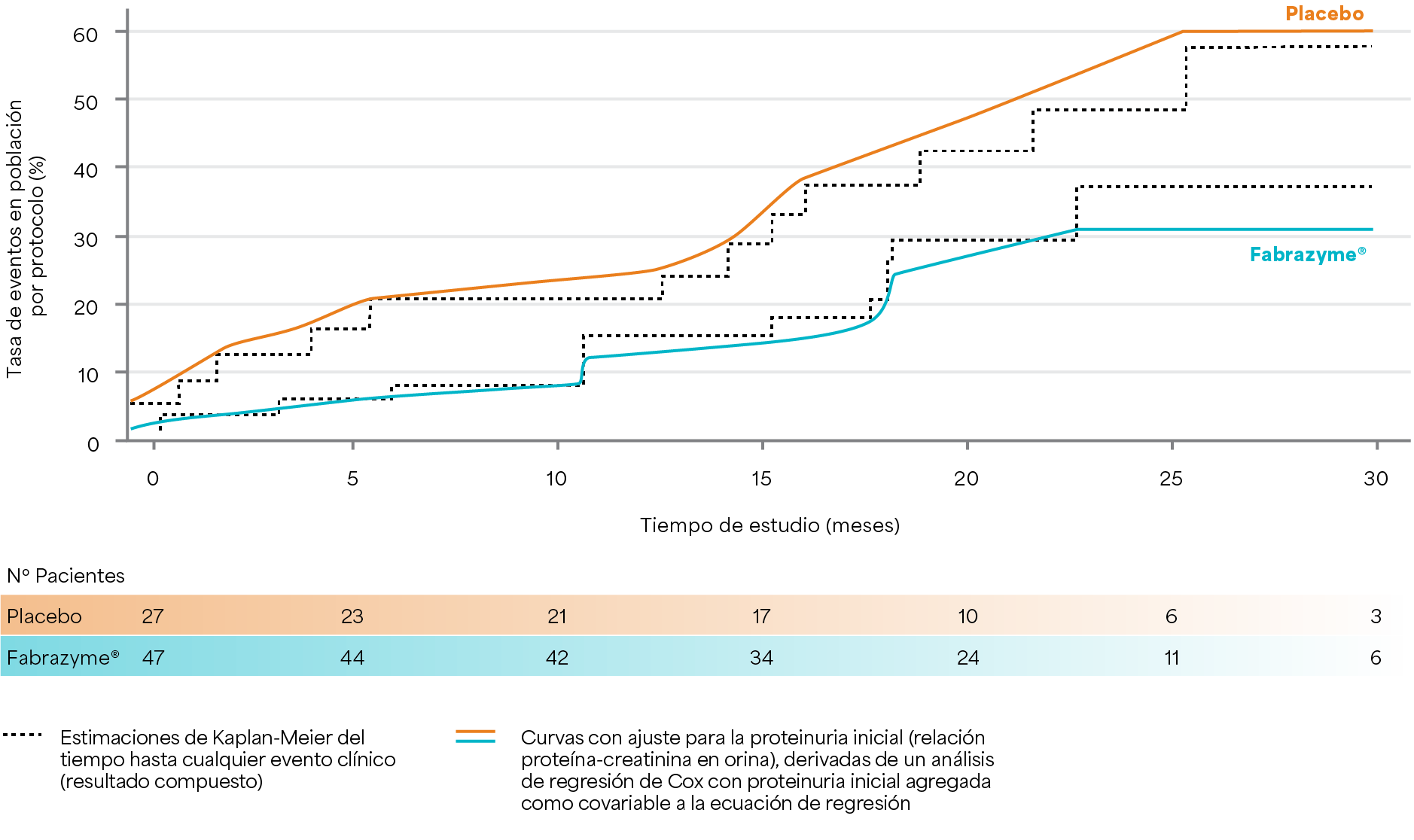
Tiempo hasta el evento para ambos grupos de tratamiento en la población por protocolo
.png)
Los resultados provienen de la población de pacientes que cumplieron con el protocolo y siguieron todas las pautas del estudio clínico después del ajuste por la proteinuria inicial2.
Banikazemi M et al. 20072.
Diseño del estudio: estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo en el que los pacientes fueron aleatorizados 2:1 para recibir agalsidasa beta (1mg/kg cada dos semanas, n=51) o placebo (n=31) cada dos semanas hasta 35 meses. El criterio de valoración principal fue el tiempo hasta el primer evento clínico (evento renal, cardíaco o cerebrovascular o muerte).2
Fabrazyme® redujo el tiempo hasta el primer evento clínico entre los pacientes tratados con Fabrazyme® versus los pacientes tratados con placebo (reducción del riesgo = 53 % de la población ITT [p = 0,06]; ** reducción del riesgo = 61% en la población adherente al protocolo [p=0,034]*).2
Protección del riñón a largo plazo
La disminución de la TFGe fue un 70,9% más lenta en pacientes tratados con Fabrazyme® que en pacientes comparables no tratados, después de ajustar por género y proteinuria*4.
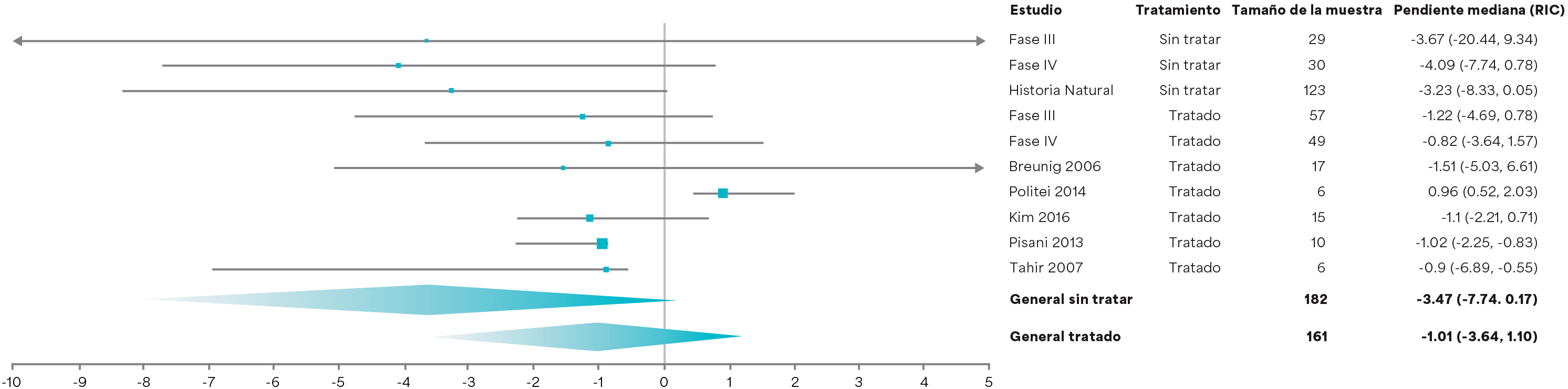
Diagrama de bosque que compara las pendientes medianas ajustadas de la TFGe en pacientes tratados con Fabrazyme® versus pacientes no tratados4 Ortiz A et al. 2021.4
Diseño del estudio: un metanálisis que determina el efecto de Fabrazyme® sobre la pérdida de TFGe en el fenotipo clásico utilizando un enfoque de modelado sólido y una amplia base de evidencia de datos a nivel de pacientes individuales que incluyen cuatro estudios de Sanofi-Genzyme y seis estudios de una revisión sistemática de literatura (N=315 pacientes).4 Se requirió que los pacientes tuvieran datos de seguimiento durante un mínimo de 12 semanas, y 67 pacientes tratados y 55 no tratados tuvieron 4 años de seguimiento.4
*Los pacientes con enfermedad de Fabry clásica tratados con Fabrazyme® experimentan un descenso de mediana de TFGe más lenta (2,46 ml/min/1,73 m2/año, IC del 95 %: [0,63–4,29]; p=0,0087) que los pacientes comparables no tratados.4
Mejora de la función cardíaca
El inicio temprano con Fabrazyme®, antes del desarrollo de fibrosis, podría ayudar a mejorar la morfología y la función cardíaca y la capacidad de ejercicio.5
Cambio en la masa del ventrículo izquierdo durante 3 años de TRE5
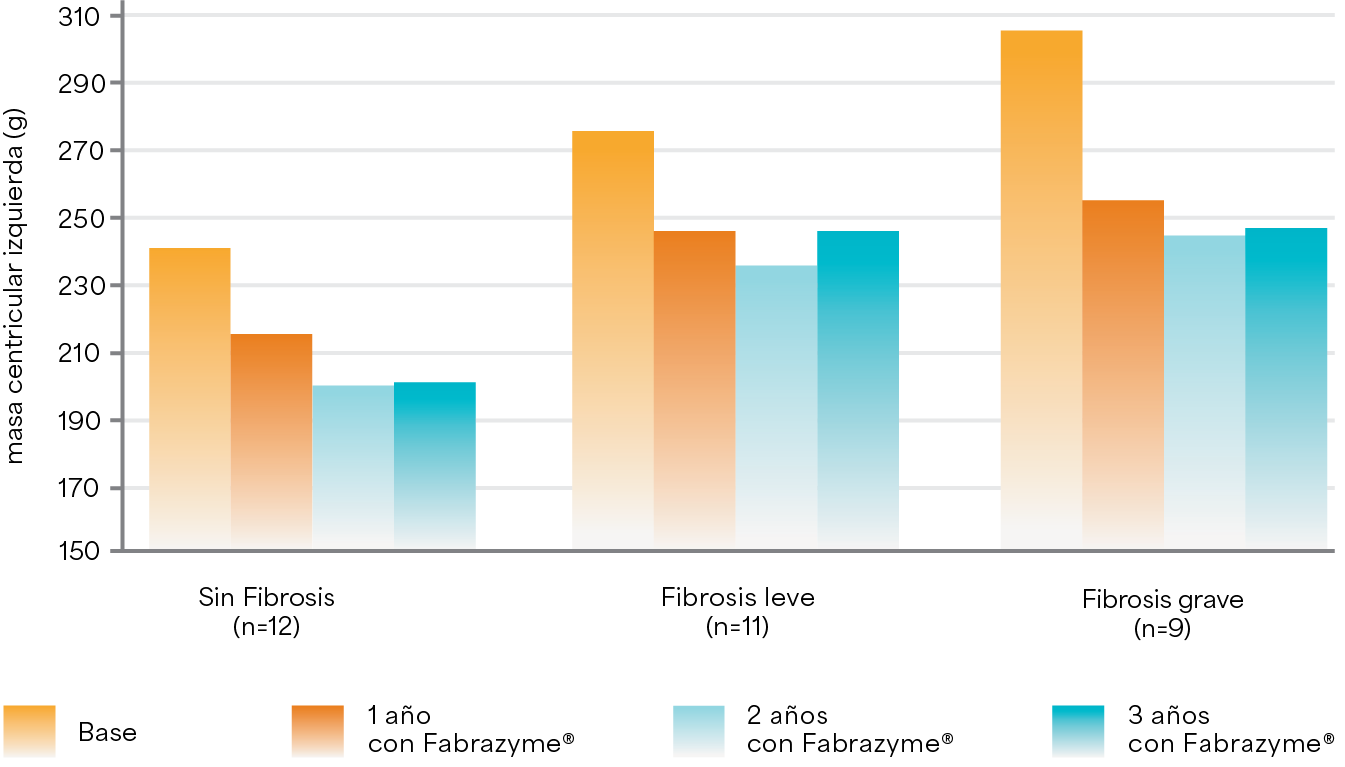
Creada a partir de la tabla 3 del artículo Weidemann F et al. 2009.5
Cambio en la capacidad de ejercicio durante 3 años de TRE5
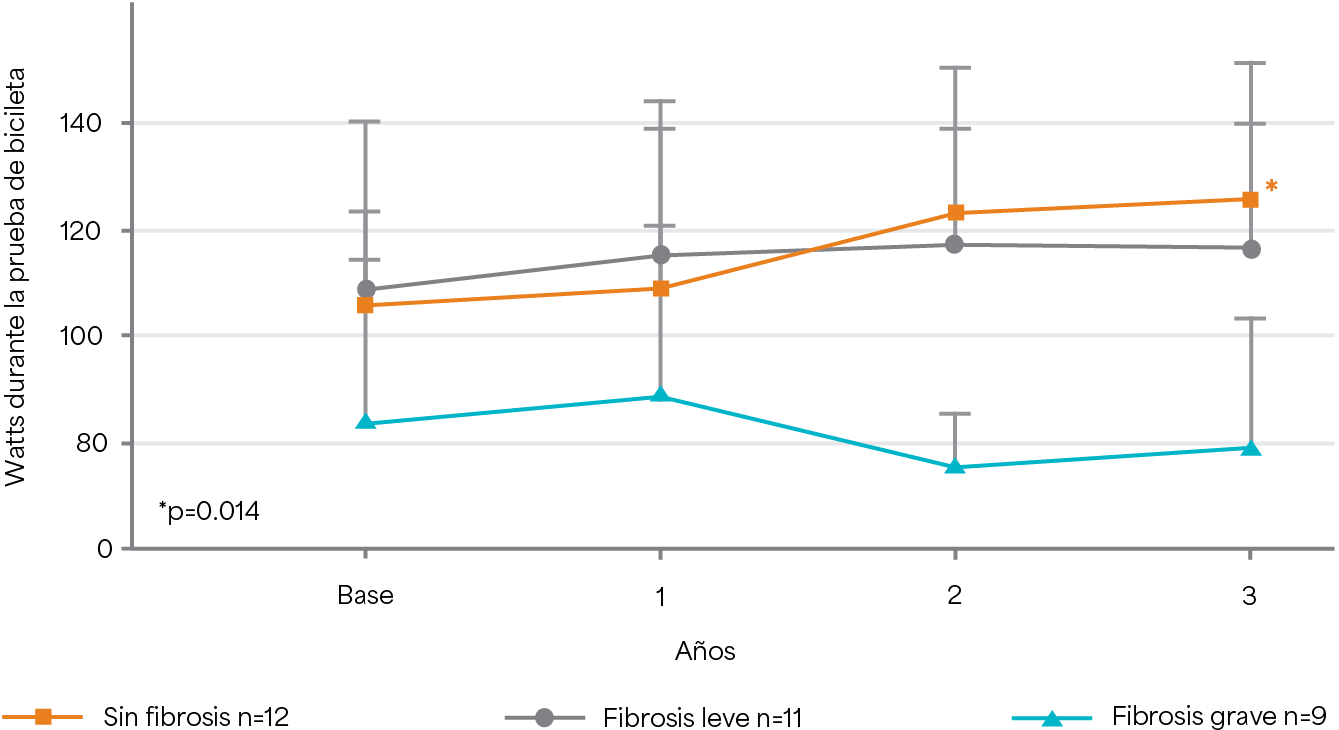
Weidemann F et al. 2009.20
Diseño del estudio: estudio observacional prospectivo de 32 pacientes que nunca habían recibido TRE (edad media 42 años) tratados con 1 mg/kg de Fabrazyme® cada dos semanas durante 3 años.5
Reducción de eventos graves
Reducción del 61% del riesgo relativo de acontecimientos clínicos renales, cardíacos y cerebrovasculares potencialmente mortales y muerte en pacientes tratados con Fabrazyme® pacientes frente a placebo*, 2.
Un porcentaje menor de pacientes tratados con Fabrazyme® (27%) experimentó eventos clínicos en comparación con placebo (42%)2.
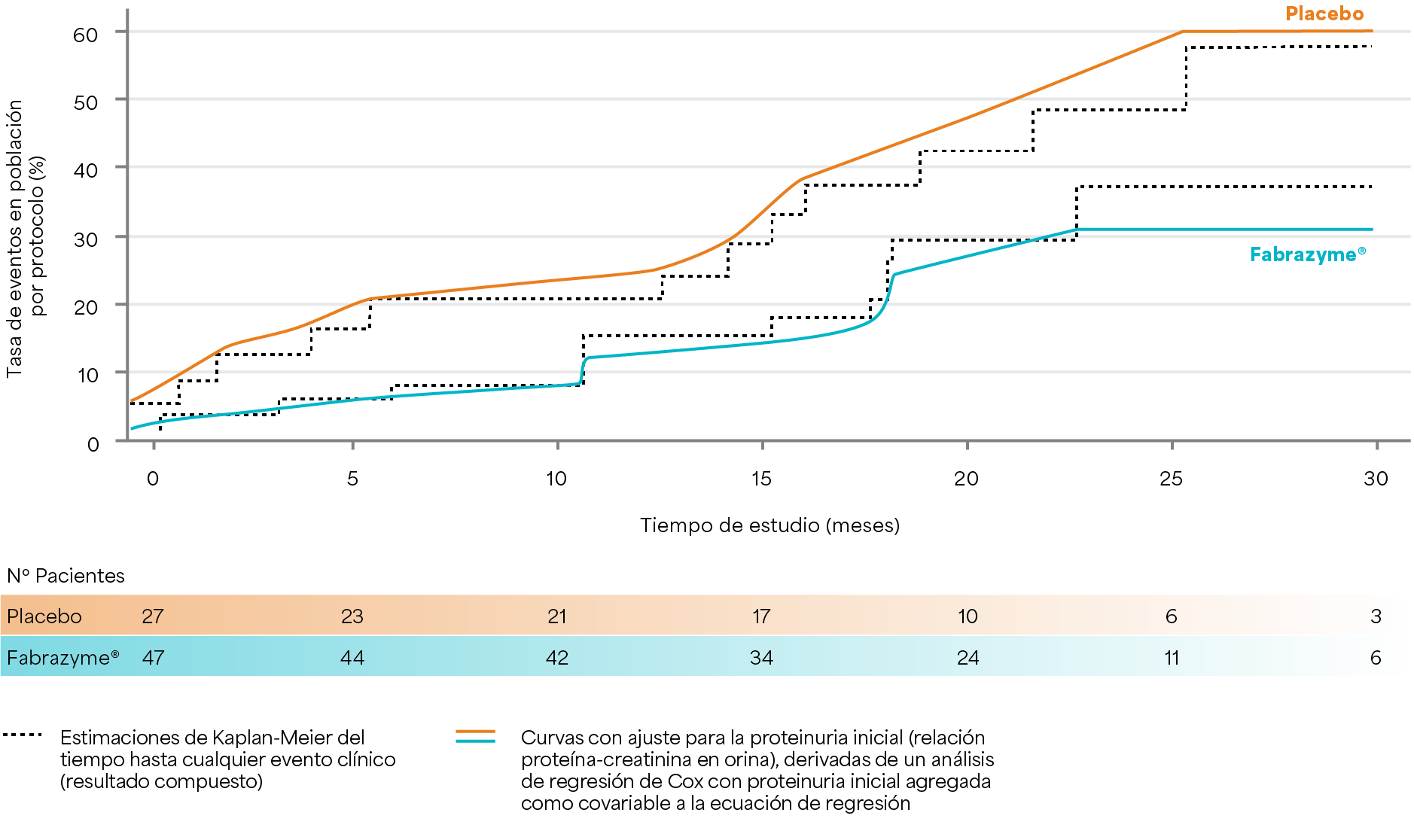
Tiempo hasta el evento para ambos grupos de tratamiento en la población por protocolo
Los resultados provienen de la población de pacientes que cumplieron con el protocolo y siguieron todas las pautas del estudio clínico después del ajuste por la proteinuria inicial2.
Banikazemi M et al. 20072.
Diseño del estudio: estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo en el que los pacientes fueron aleatorizados 2:1 para recibir agalsidasa beta (1mg/kg cada dos semanas, n=51) o placebo (n=31) cada dos semanas hasta 35 meses. El criterio de valoración principal fue el tiempo hasta el primer evento clínico (evento renal, cardíaco o cerebrovascular o muerte).2
Fabrazyme® redujo el tiempo hasta el primer evento clínico entre los pacientes tratados con Fabrazyme® versus los pacientes tratados con placebo (reducción del riesgo = 53 % de la población ITT [p = 0,06]; ** reducción del riesgo = 61% en la población adherente al protocolo [p=0,034]*).2
Protección del riñón a largo plazo
La disminución de la TFGe fue un 70,9% más lenta en pacientes tratados con Fabrazyme® que en pacientes comparables no tratados, después de ajustar por género y proteinuria*4.
Diagrama de bosque que compara las pendientes medianas ajustadas de la TFGe en pacientes tratados con Fabrazyme® versus pacientes no tratados4 Ortiz A et al. 2021.4
Diseño del estudio: un metanálisis que determina el efecto de Fabrazyme® sobre la pérdida de TFGe en el fenotipo clásico utilizando un enfoque de modelado sólido y una amplia base de evidencia de datos a nivel de pacientes individuales que incluyen cuatro estudios de Sanofi-Genzyme y seis estudios de una revisión sistemática de literatura (N=315 pacientes).4 Se requirió que los pacientes tuvieran datos de seguimiento durante un mínimo de 12 semanas, y 67 pacientes tratados y 55 no tratados tuvieron 4 años de seguimiento.4
*Los pacientes con enfermedad de Fabry clásica tratados con Fabrazyme® experimentan un descenso de mediana de TFGe más lenta (2,46 ml/min/1,73 m2/año, IC del 95 %: [0,63–4,29]; p=0,0087) que los pacientes comparables no tratados.4
Mejora de la función cardíaca
El inicio temprano con Fabrazyme®, antes del desarrollo de fibrosis, podría ayudar a mejorar la morfología y la función cardíaca y la capacidad de ejercicio.5
Cambio en la masa del ventrículo izquierdo durante 3 años de TRE5
Creada a partir de la tabla 3 del artículo Weidemann F et al. 2009.5
Cambio en la capacidad de ejercicio durante 3 años de TRE5
Weidemann F et al. 2009.20
Diseño del estudio: estudio observacional prospectivo de 32 pacientes que nunca habían recibido TRE (edad media 42 años) tratados con 1 mg/kg de Fabrazyme® cada dos semanas durante 3 años.5
Eficacia rápida y sostenida
.png)
El 94% de los pacientes con Fabry siguen vivos después de 10 años de tratamiento con Fabrazyme®*,3.
.png)
El 81% de los pacientes no experimentó ningún evento clínico renal, cardíaco o cerebrovascular grave durante 10 años de tratamiento con Fabrazyme**,3.
*Se evaluaron datos basados en un análisis de pacientes con enfermedad de Fabry clásica en el ensayo clínico de fase 3 y el estudio de extensión, y el registro de Fabry.3
**Los eventos clínicos graves se definieron como diálisis crónica, trasplante de riñón, infarto de miocardio, insuficiencia cardíaca congestiva, procedimientos cardíacos mayores, accidente cerebrovascular y muerte.3
Aclaramiento rápido y sostenido de GL-3 en riñón, corazón y piel6,7
Fabrazyme® eliminó GL-3 en riñón, corazón y piel en 6 meses8
Fabrazyme® redujo la GL-3 plasmática a niveles normales (7,03 μg/ml) en los 6 meses posteriores al inicio del tratamiento y los mantuvo durante 54 meses.8
Diseño del estudio: Estudio aleatorizado, doble ciego y controlado con placebo de 58 pacientes con diagnóstico de enfermedad de Fabry, de edades comprendidas entre 16 y 61 años, incluidos 56 hombres y 2 mujeres, que no habían recibido TRE.6
Los pacientes fueron aleatorizados 1:1 para recibir 1 mg/kg de Fabrazyme® cada dos semanas o placebo durante 5 meses (20 semanas).6
Los 58 pacientes se inscribieron en un estudio de extensión abierto en el que recibieron Fabrazyme® 1 mg/kg quincenalmente hasta 54 meses adicionales.8
Germain D et al. 2007.8
Seguridad
.jpg)
El primer tratamiento en demostrar una tasa sustancialmente menor de acontecimientos renales, cardíacos, cerebrovasculares y muertes en la población por protocolo frente a la tratada con placebo, en pacientes con enfermedad de Fabry.1-3
Fabrazyme está indicado como terapia de sustitución enzimática a largo plazo en pacientes con diagnóstico confirmado de enfermedad de Fabry (déficit de α-galactosidasa A).
Fabrazyme está indicado en adultos, adolescentes y niños de 8 años o mayores.
Biomarcador Liso-GL3
La dosis importa
Liso-GL3 es un marcador diagnóstico que contribuye a la patogenia de la enfermedad de Fabry9-14 y constituye un factor de riesgo asociado a eventos clínicos graves.15
El Consenso Europeo de objetivos terapéuticos en enfermedad de Fabry, recomienda mantener el nivel de liso-GL3 lo más bajo posible.13
Los niveles de liso-GL3 y de GL3 se redujeron tras cambiar de agalsidasa alfa 0,2 mg/kg a agalsidasa beta 1 mg/kg, indicando que agalsidasa beta tiene mayor efecto que alfa sobre estos marcadores y apoyando el uso de agalsidasa beta 1mg/kg como TRE en enfermedad de Fabry.16-18
Reducción de liso-GL3 al cambiar de agalsidasa alfa 0,2 mg/kg a agalsidasa beta 1 mg/kg16
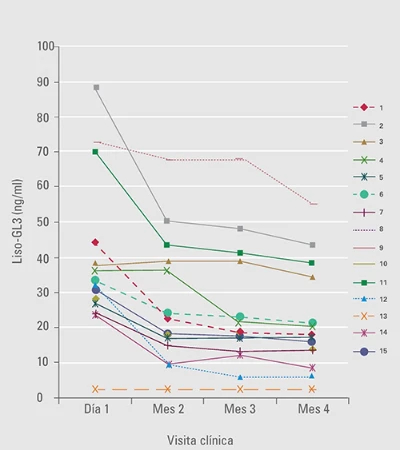.jpg)
La disminución de liso-GL3 fue mayor con agalsidasa beta que con agalsidasa alfa17.
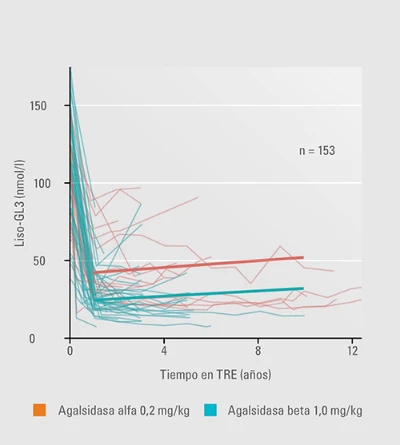
Liso-GL3 aumenta al cambiar de agalsidasa beta a alfa y vuelve a disminuir en el re-switch18.
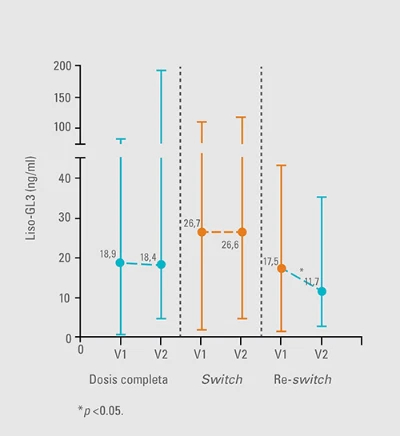
El seguimiento importa
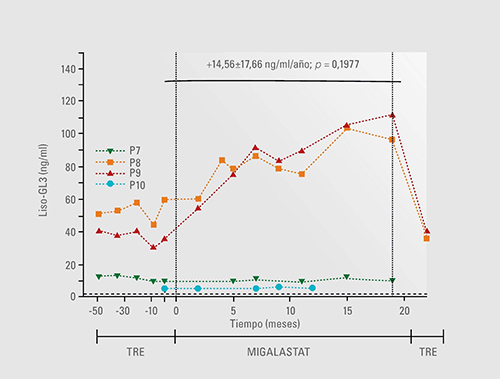
Liso-GL3 aumentó en 2 pacientes con una variante (p.L294S) susceptible in vitro a Migalastat y volvió a disminuir en el re-switch a TRE19.
.jpeg)
Dos pacientes varones con una mutación susceptible in vitro a Migalastat, experimentaron un aumento de los niveles de liso-GL3 después del cambio de TRE a Migalastat. Esto condujo a una albuminuria severa y requirió volver a cambiar a TRE. Liso-GL3 volvió a disminuir tras cambiar de nuevo a TRE.19
Los pacientes en tratamiento con Migalastat que tengan una actividad baja de galactosidasa y un nivel alto de liso-GL3, podrían no estar beneficiándose de este tratamiento, a pesar de que la mutación sea susceptible in vitro.19
Se sugiere que se debe a que algunas variantes susceptibles in vitro no responden como se esperaba. Esto indica que es necesaria una monitorización cuidadosa de los pacientes que inicien terapia con chaperona oral.20
La dosis importa
Liso-GL3 es un marcador diagnóstico que contribuye a la patogenia de la enfermedad de Fabry9-14 y constituye un factor de riesgo asociado a eventos clínicos graves.15
El Consenso Europeo de objetivos terapéuticos en enfermedad de Fabry, recomienda mantener el nivel de liso-GL3 lo más bajo posible.13
Los niveles de liso-GL3 y de GL3 se redujeron tras cambiar de agalsidasa alfa 0,2 mg/kg a agalsidasa beta 1 mg/kg, indicando que agalsidasa beta tiene mayor efecto que alfa sobre estos marcadores y apoyando el uso de agalsidasa beta 1mg/kg como TRE en enfermedad de Fabry.16-18
Reducción de liso-GL3 al cambiar de agalsidasa alfa 0,2 mg/kg a agalsidasa beta 1 mg/kg16
La disminución de liso-GL3 fue mayor con agalsidasa beta que con agalsidasa alfa17.
Liso-GL3 aumenta al cambiar de agalsidasa beta a alfa y vuelve a disminuir en el re-switch18.
El seguimiento importa
Liso-GL3 aumentó en 2 pacientes con una variante (p.L294S) susceptible in vitro a Migalastat y volvió a disminuir en el re-switch a TRE19.
Dos pacientes varones con una mutación susceptible in vitro a Migalastat, experimentaron un aumento de los niveles de liso-GL3 después del cambio de TRE a Migalastat. Esto condujo a una albuminuria severa y requirió volver a cambiar a TRE. Liso-GL3 volvió a disminuir tras cambiar de nuevo a TRE.19
Los pacientes en tratamiento con Migalastat que tengan una actividad baja de galactosidasa y un nivel alto de liso-GL3, podrían no estar beneficiándose de este tratamiento, a pesar de que la mutación sea susceptible in vitro.19
Se sugiere que se debe a que algunas variantes susceptibles in vitro no responden como se esperaba. Esto indica que es necesaria una monitorización cuidadosa de los pacientes que inicien terapia con chaperona oral.20
Mecanismo de acción

Contenido mínimo de Fabrazyme®
PRESENTACIÓN, PRECIO Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN:
Fabrazyme 35 mg polvo para concentrado para solución para perfusión, 1 vial (CN 961631.1): PVP: 3.426,35 €, PVP IVA: 3.563,40 €. Medicamento sujeto a prescripción médica. Financiado por el SNS. Uso hospitalario.
Fabrazyme 5 mg polvo para concentrado para solución para perfusión, 1 vial (CN 758128.4): PVP: 558,51 €, PVP IVA: 580,85 €. Medicamento sujeto a prescripción médica. Financiado por el SNS. Uso hospitalario.
Fecha de la revisión del texto: Marzo 2024
1. NOMBRE DEL MEDICAMENTO
Fabrazyme 35 mg polvo para concentrado para solución para perfusión
Fabrazyme 5 mg polvo para concentrado para solución para perfusión
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Fabrazyme 35 mg polvo para concentrado para solución para perfusión
Cada vial de Fabrazyme contiene un valor nominal de 35 mg de agalsidasa beta. Una vez reconstituido con 7,2 ml de agua para preparaciones inyectables, cada vial de Fabrazyme contiene 5 mg/ml (35 mg/7 ml) de agalsidasa beta. La solución reconstituida se tiene que diluir a posteriori (ver sección 6.6 de Ficha Técnica).
Fabrazyme 5 mg polvo para concentrado para solución para perfusión
Cada vial de Fabrazyme contiene un valor nominal de 5 mg de agalsidasa beta. Una vez reconstituido con 1,1 ml de agua para preparaciones inyectables, cada vial de Fabrazyme contiene 5 mg/ml de agalsidasa beta. La solución reconstituida se tiene que diluir a posteriori (ver sección 6.6 de Ficha Técnica).
La agalsidasa beta es una forma recombinante de la -galactosidasa A humana y se obtiene mediante tecnología de ADN recombinante a partir de un cultivo de células de mamífero procedentes de ovario de hámster chino (CHO). La secuencia de aminoácidos de la forma recombinante así como la secuencia nucleótida que la codificó son idénticas a las de la forma natural de la -galactosidasa A.
Para consultar la lista completa de excipientes, ver sección 6.1 de Ficha Técnica.
3. FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión. Polvo liofilizado de color blanco a blanquecino.
IC: intervalo de confianza. TFGe: tasa de filtrado glomerular estimada. GL-3: globotriaosilceramida. ITT: intención de tratar. RIQ: rango intercuartil.
Referencias
- Ficha Técnica de Fabrazyme® (agalsidase beta).
- Banikazemi M et al. Ann Intern Med 2007; 146: 77–86.
- Germain D et al. J Med Genet 2015; 52(5): 353–358.
- Ortiz A et al. Clinical Kidney Journal 2021; 14 (4): 1136–1146.
- Weidemann F et al. Circulation 2009; 119(4): 524–529.
- Eng CM et al. N Engl J Med 2001; 345(1): 9–16.
- Thurberg B et al. Circulation 2009; 119(19): 2561–2567.
- Germain D et al. J Am Soc Nephrol 2007; 18(5): 1547–1557.
- Smid BE, et al. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J Med Genet. 2015;52(4):262–8.
- Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416-27.
- Rombach SM, et al. Plasma globotriaosylsphingosine: Diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta. 2010;1802:741-8.
- Rozenfeld P, et al. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122(3):19-27.
- Wanner C, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189-203.
- Liao H-C, et al. Plasma globotriaosylsphingosine could be a biomarker for Fabry disease with a Chinese hotspot late-onset mutation. Clin Chim Acta. 2013;426:114-20.
- Nowak A, et al. Lyso-Gb3 associates with adverse long-term outcome in patients with Fabry disease. J Med Genet 2022;59(3):287-93.
- Goker-Alpan O, et al. Reduction of plasma globotriaosylsphingosine levels after switching from agalsidase alfa to agalsidase beta as enzyme replacement therapy for Fabry disease. JIMD Rep. 2016;25:95-106.
- Arends M, et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: an international cohort study. J Med Genet. 2018;55(5):351-8.
- Krämer J, et al. Fabry disease under enzyme replacement therapy—new insights in efficacy of different dosages. Nephrol Dial Transplant. 2018;33(8):1362-72.
- Lenders M, et al. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J Med Genet. 2019;56(8):548-56.
- Germain DP, et al. Consenso de expertos sobre recomendaciones de práctica clínica y manejo de pacientes con enfermedad de Fabry clásica. Mol Genet Metab. 2022;137(1-2):49-61.
MAT-ES-2303248 V1 Marzo 2024