- Artículo
- Fuente: Campus Sanofi
- 15 abr 2023
Diagnóstico y pruebas de la enfermedad de Gaucher
Diagnóstico diferencial
La capacidad de diferenciar correctamente la enfermedad de Gaucher de otras enfermedades es crucial para minimizar el retraso en el diagnóstico y optimizar las respuestas de los pacientes. Sin embargo, diagnosticar la enfermedad de Gaucher puede resultan una tarea difícil debido a su rareza, a su evolución clínica impredecible y variable, y al hecho de que muchos de sus signos y síntomas clínicos pueden atribuirse a otras enfermedades. A pesar de que se dispone de pruebas de diagnóstico definitivas que incluyen pruebas de actividad enzimática y análisis de ADN, son bastante comunes los retrasos considerables a la hora del diagnóstico.1
Cuando un paciente presenta la enfermedad de Gaucher, inicialmente pueden sospecharse de otras enfermedades. Entre ellos:1
-
Leucemia.
-
Linfoma.
-
Trastornos hemorrágicos.
-
Mieloma múltiple.
La esplenomegalia es un signo de presentación clave de la enfermedad de Gaucher que se observa en la mayoría de los pacientes de Gaucher. Otros signos y síntomas son particularmente indicativos de la enfermedad de Gaucher y debe realizarse un seguimiento inmediato de los mismos:1
-
Los médicos deben considerar el diagnóstico de enfermedad de Gaucher tipo 1 (no neuronopática) en cualquier paciente que presente esplenomegalia idiopática, con o sin manifestaciones óseas, hepatomegalia y diátesis hemorrágica.
-
Los niños que presenten hepatoesplenomegalia y síntomas neurológicos también deben ser evaluados por si padecen enfermedad de Gaucher tipo 2 o 3.
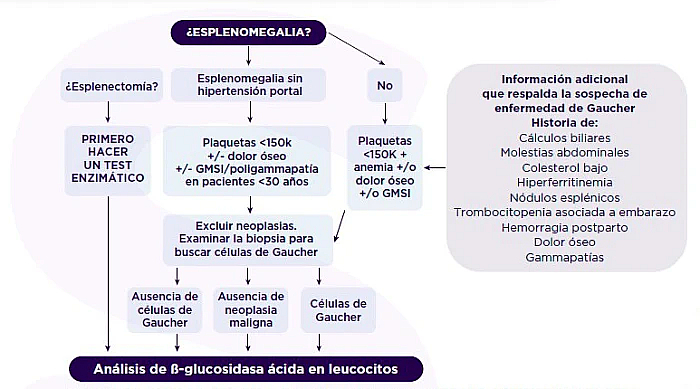
Recientemente, un grupo internacional ha publicado una serie de útiles algoritmos de diagnóstico que muestran las frecuencias de las enfermedades relacionadas con la esplenomegalia.1
Diagnóstico de enfermedad de Gaucher en pacientes sin ascendencia de etnia asquenazí.1
Enfermededad de Gaucher 1: 40 000-10 000 neoplasias malignas hematológicas 1:2500 1
Poblaciones de alto riesgo
Las poblaciones de alto riesgo en las que se deberían realizar más pruebas diagnósticas para detectar la enfermedad de Gaucher incluyen a los hermanos de las personas que padecen la enfermedad, así como a aquellos con ascendencia judía asquenazí. Puede resultar conveniente realizar un cribado poblacional en algunas poblaciones como Norbotnia, Suecia, o en las poblaciones árabes de las regiones de Jenin y Gaza en Palestina, donde hay una alta prevalencia del tipo 3 de la enfermedad.1

Pruebas diagnósticas y de confirmación
La enfermedad de Gaucher es una enfermedad rara que presenta signos y síntomas que pueden confundirse con los de muchas otras afecciones. Sin embargo, existen una serie de pruebas que permiten emitir el diagnóstico definitivo de la enfermedad.1
Análisis enzimático
La enfermedad de Gaucher está causada por una reducción significativa de la enzima ß-glucosidasa ácida. La actividad enzimática de la ß-glucosidasa ácida puede medirse utilizando una muestra de sangre seca (dry blood spot, DBS). Sin embargo, el método de referencia para el diagnóstico de la enfermedad de Gaucher es la demostración de la actividad deficiente de la enzima ß-glucosidasa ácida en leucocitos periféricos, fibroblastos cultivados o en células del líquido amniótico. Los análisis enzimáticos no distinguen entre los diferentes tipos de enfermedad de Gaucher.1

Diagnóstico molecular
La prueba de ADN puede utilizarse para ayudar a diagnosticar y detectar el estado de portador. Aunque ya se han identificado actualmente numerosas mutaciones asociadas a la enfermedad de Gaucher, un resultado negativo no garantiza necesariamente la ausencia de un alelo de la enfermedad de Gaucher. Sin embargo, se ha demostrado la presencia de algunas asociaciones, sobre todo en la población judía asquenazí. En este grupo, aproximadamente 1 de cada 17 personas es portadora de la mutación en el gen de la ß-glucosidasa ácida 1 (GBA1). Dos mutaciones son las responsables de hasta un 80‑90 % de las mutaciones observadas en pacientes judíos asquenazíes con la enfermedad de Gaucher: N370S y 84GG. Las mutaciones L444P y N370S representan más de la mitad de las mutaciones en pacientes blancos no judíos.1
Pruebas histológicas
El método clásico para diagnosticar la enfermedad de Gaucher era la identificación de células de Gaucher cargadas de lípidos en un aspirado medular, una biopsia hepática o una esplenectomía. Sin embargo, se han descrito células muy similares (llamadas células pseudo-Gaucher) relacionadas con otras enfermedades entre las que se incluyen:1
- leucemia granulocítica crónica;
- leucemia linfocítica aguda;
- linfomas;
- talasemia;
- mieloma múltiple;
- enfermedad de Hodgkin.
Las técnicas bioquímicas y moleculares son más específicas y menos invasivas por lo que actualmente se desaconseja la evaluación histológicade rutina.1
Marcadores bioquímicos
El plasma de los pacientes con la enfermedad de Gaucher presenta niveles elevados de diversas enzimas específicas de los macrófagos y algunas pueden utilizarse como biomarcadores en la monitorización periódica. El hallazgo de niveles elevados de uno o más de estos marcadores nunca es suficiente para diagnosticar la enfermedad de Gaucher, ya que estos marcadores también pueden presentar niveles elevados en otros trastornos. Estos biomarcadores incluyen:1
- Quitotriosidasa
- Enzima convertidora de angiotensina
- Fosfatasa ácida tartrato-resistente (tartrate-resistant acid phosphatase, TRAP)
- LysoGL1
Además, en pacientes con la enfermedad de Gaucher también suelen observarse niveles elevados de ferritina, niveles bajos de vitamina B12 e hipocolesterolemia. No obstante, los biomarcadores mencionados en esta sección no son suficientes ni necesarios para emitir un diagnóstico definitivo de Gaucher dada su vinculación con otros muchos trastornos.1
Diagnóstico familiar
Al tratarse de una enfermedad que se transmite en las familias, si una persona padece la enfermedad de Gaucher, en ese caso es muy probable que otros familiares presenten el gen defectuoso. Es muy importante que los hermanos de alguien que tenga la enfermedad de Gaucher se sometan a una prueba por si también padeciesen la enfermedad. Esta prueba está disponible en centros especializados y se suele conocer como “cribado familiar” o “cribado de hermanos”.1
Predecir la gravedad de la enfermedad en un hijo es más fácil cuando los padres ya tienen otro hijo con la enfermedad de Gaucher al existir un considerable grado de concordancia entre hermanos. En ocasiones, el grado de manifestación de la enfermedad es completamente diferente entre hermanos, pero esto es menos frecuente.1
Si no hay hijos con la enfermedad de Gaucher en la familia, el genetista se sirve del genotipo de los progenitores, el cual puede proporcionar información útil. Por ejemplo, si ambos progenitores son heterocigotos para la mutación N370S, entonces pueden tener la tranquilidad de que el futuro hijo no padecerá enfermedad neuronopática.1
Continúa explorando otros contenidos:

Presentación Clínica y diagnóstico
Conoce más sobre la presentación de esta enfermedad

¿ASMD o GAUCHER?
Aprende a diferenciar estas enfermedades con GlorIA
Presentación Clínica y diagnóstico
Conoce más sobre la presentación de esta enfermedad
¿ASMD o GAUCHER?
Aprende a diferenciar estas enfermedades con GlorIA


Referencias
- Grabowski GA, Petsko GA, Kolodny EH. Gaucher Disease. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; Accessed July 16, 2020.
MAT-ES-2301496 V2 Septiembre 2025