- Artículo
- Fuente: Campus Sanofi
- 22 mar 2024
Enfermedad de Gaucher
¿Conoces la enfermedad de Gaucher? Podrías tener un caso delante y no saberlo.
Es una enfermedad genética causada por el déficit de la enzima β-glucosidasa ácida1.
Esto provoca acumulación de lípidos en el bazo, hígado, médula ósea…1. El tipo 1 es el más común1: sin afectación neurológica, pero con manifestaciones viscerales, hematológicas y afectación ósea1 . Puede aparecer en la infancia o en adultos.
Haz clic para conocer mejor esta enfermedad rara y mejorar tu sospecha clínica desde la primera consulta.
¿Conoces la enfermedad de Gaucher? Podrías tener un caso delante y no saberlo.
Es una enfermedad genética causada por el déficit de la enzima β-glucosidasa ácida1.
Esto provoca acumulación de lípidos en el bazo, hígado, médula ósea…1. El tipo 1 es el más común1: sin afectación neurológica, pero con manifestaciones viscerales, hematológicas y afectación ósea1 . Puede aparecer en la infancia o en adultos.
Haz clic para conocer mejor esta enfermedad rara y mejorar tu sospecha clínica desde la primera consulta.
Continúa navegando estos contenidos para descifrar la enfermedad de Gaucher junto a GlorIA:
Historia y patogenia de la enfermedad
La enfermedad fue descrita por primera vez por un estudiante de medicina francés Philippe Gaucher en 1882 que describió el caso de una mujer joven que presentaba agrandamiento del bazo y células congestionadas características. Más de 50 años después, Aghion identificó que los pacientes con esta enfermedad acumulaban un esfingolípido denominado glucosilceramida, pero no fue hasta 1965 cuando Brady y sus colaboradores demostraron que la enfermedad de Gaucher estaba causada por la actividad deficiente de la enzima ß-glucosidasa ácida.1
La enfermedad de Gaucher es un trastorno de depósito lisosomal causado por una mutación en el gen responsable de la producción de la enzima ß-glucosidasa ácida. Hasta la fecha se han identificado más de 300 alelos mutantes.1
Normalmente, la β-glucosidasa ácida degrada los glucoesfingolípidos derivados del recambio fisiológico de membranas, particularmente de las células sanguíneas. Sin embargo, la mutación genética disminuye la actividad de la ß-glucosidasa ácida haciéndola insuficiente para evitar la acumulación de un glucoesfingolípido denominado glucosilceramida en los lisosomas de las células, principalmente en los macrófagos; los macrófagos cargados de glucosilceramida, llamados células de Gaucher, se acumulan en los órganos. Esa acumulación de células de Gaucher provoca una secuencia de acontecimientos fisiopatológicos, incluida la aparición de un estado inflamatorio e hipermetabólico crónico.1
Se trata de una enfermedad multisistémica que varía significativamente en cuanto a sus manifestaciones clínicas, intensidad y evolución. La deficiencia parcial de ß-glucosidasa ácida se asocia con afectación a nivel del hígado, el bazo, la médula ósea y el pulmón. La deficiencia severa de la enzima también se asocia con manifestaciones neurológicas.1
Clasificación de la enfermedad
Por lo general, la enfermedad de Gaucher se clasifica en tres tipos en función de la ausencia (tipo 1) o presencia (tipos 2 y 3) de afectación neurológica:1,2
- Tipo 1 (no neuronopática): presenta una gran variabilidad en cuanto a sus manifestaciones clínicas, intensidad y evolución, pero se distingue de los otros dos tipos por la ausencia de afectación primaria del sistema nervioso central. En algunas personas, los síntomas comienzan temprano en la infancia y empeoran con el tiempo, mientras que en otras, puede que los primeros síntomas solo se hagan perceptibles en la edad adulta.
- Tipo 2 (neuronopática aguda): por lo general se hace evidente en los primeros meses de vida e incluye síntomas neurológicos graves. Los niños no suelen sobrevivir más allá de los dos años de edad.
- Tipo 3 (neuronopática crónica): se caracteriza por ser una enfermedad neurológica de progresión lenta y puede confundirse con el tipo 1 de la enfermedad en sus primeras etapas. Los pacientes que alcanzan la adolescencia pueden vivir hasta la edad adulta.
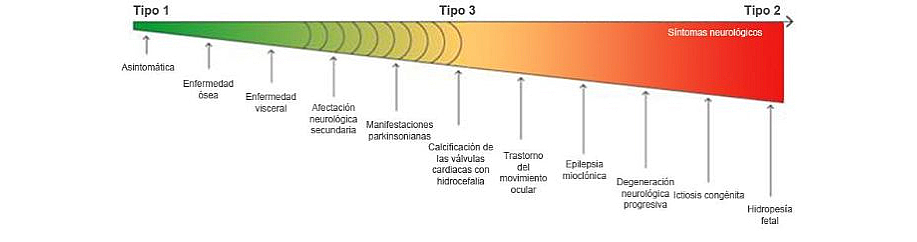
Clasificar los diferentes tipos de enfermedad de Gaucher es valioso desde el punto de vista de la evaluación de las opciones terapéuticas y como base para el asesoramiento genético, pero existe cada vez un mayor consenso según el cual la enfermedad de Gaucher debería verse como un continuum fenotípico con diversas manifestaciones que abarcan síntomas neurológicos de grado leve a grave.1,2
Enfermedad de Gaucher: un continuum fenotípico1,2
.2024-04-22-14-59-54.jpg)
La división establecida en las categorías clásicas de los tipos 1, 2 y 3 es difusa a lo largo del continuum de la enfermedad. Adaptado de Sidransky E, Gaucher disease: complexity in a “simple” disorder.1
Progresión y pronóstico
La enfermedad de Gaucher tipo 1 se caracteriza por una amplia variabilidad y diversidad fenotípica. La edad de aparición y diagnóstico también difieren ampliamente de una persona a otra, aunque al nacer los bebés son asintomáticos. En casos graves, la visceromegalia puede hacerse evidente después del primer año de vida. Con frecuencia, la enfermedad en adultos presenta una progresión de la enfermedad muy lenta. Sin embargo, estas personas precisan una evaluación cuidadosa y exhaustiva, ya que no se puede descartar la posibilidad de osteopatía severa cuando las manifestaciones hematológicas/viscerales no están claramente presentes. La esperanza de vida varía mucho, pero puede ser entre normal y casi normal.1,2
La enfermedad de Gaucher tipo 2 tiene un pronóstico grave con muerte prematura debido al empeoramiento de los síntomas neurológicos. La evolución clínica general es homogénea aunque existe cierta heterogeneidad, especialmente en la edad de aparición; no obstante, la mayoría de los bebés muere en los dos primeros años de vida.1,2
La enfermedad de Gaucher tipo 3 presenta, entre otros, síntomas neuronopáticos de moderados a graves que normalmente comienzan en la infancia y progresan lentamente.1,2
Presentación clínica
La enfermedad de Gaucher es una enfermedad multisistémica con una amplia variación fenotípica de una persona a otra. En muchas ocasiones, los síntomas heterogéneos observados hacen que su diagnóstico suponga todo un desafío. Sin embargo, existen algunos síntomas y signos observados con más frecuencia en los diferentes tipos de enfermedad de Gaucher.1,2
Enfermedad de Gaucher tipo 1
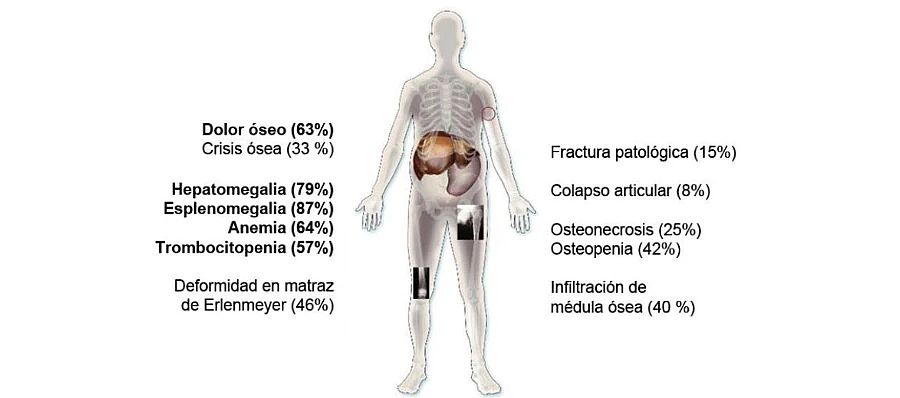
La esplenomegalia es una de las manifestaciones más frecuentes en la enfermedad tipo 1 y muchas veces puede ser el hallazgo inicial reconocido cuando un niño es pequeño. Sin embargo, estos pacientes también pueden desarrollar enfermedad ósea, posiblemente dando lugar a una osteoporosis generalizada complicada por frecuentes fracturas patológicas. Las anomalías óseas son también muy frecuentes ya que casi todos los pacientes adultos presentan complicaciones óseas. Existen una gran variedad de signos y síntomas de diferente intensidad como por ejemplo los siguientes: 1,2
Viscerales
- Esplenomegalia
- Hepatomegalia
Hematológicos
- Facilidad para los hematomas y tendencia a las hemorragias asociado a la trombocitopenia
- Anemia
Óseos
- Dolor óseo/crisis óseas
- Retraso del crecimiento (niños y adolescentes)
- Necrosis avascular
- Fracturas patológicas
- Osteopenia
- Infiltración de médula ósea
Calidad de vida
- Fatiga
- Alteración de las funciones sociales y físicas
La enfermedad tipo 1 se caracteriza por la ausencia de afectación primaria del sistema nervioso central.1,2
Esplenomegalia: uno de los signos más frecuente de la enfermedad de Gaucher
Enfermedad de Gaucher tipo 2
La enfermedad tipo 2 también presenta diversos fenotipos. Este tipo neuronopático agudo es la forma más grave de la enfermedad e incluye varios síntomas y signos relacionados con la enfermedad tipo 1 y una serie de manifestaciones neurológicas adicionales que generalmente conllevan una muerte temprana. Las características de la enfermedad tipo 2 incluyen:
- Parálisis supranuclear de la mirada
- Opistótonos
- Hidropesía fetal
- Alteración cutánea similar a la ictiosis
- Complicaciones oculares como el estrabismo
- Trismo
Enfermedad de Gaucher tipo 3
Generalmente, se considera que la gravedad de la enfermedad tipo 3 (neuronopática crónica) se encuentra entre la de los tipos 1 y 2, aunque también existen formas menos graves de la tipo 3. Además de diversos síntomas y signos asociados al tipo 1, la enfermedad tipo 3 también presenta afectación neurológica (con una aparición tardía y relativamente menos grave que en el tipo 2):
- Parálisis supranuclear de la mirada
- Ataxia
Continúa explorando otros contenidos:

Presentación Clínica y diagnóstico
Cómo diagnosticar Gaucher

Biomarcadores y Afectación Ósea
Marcadores bioquímicos para ayudarte al seguimiento en Gaucher

Solicitud DBS y Contacto Gaucher
¿Sospecha de Gaucher? Solicita test y contacto

¿ASMD o Gaucher?
Descifra estas dos enfermedades raras y sus diferencias
Presentación Clínica y diagnóstico
Cómo diagnosticar Gaucher
Biomarcadores y Afectación Ósea
Marcadores bioquímicos para ayudarte al seguimiento en Gaucher
Solicitud DBS y Contacto Gaucher
¿Sospecha de Gaucher? Solicita test y contacto
¿ASMD o Gaucher?
Descifra estas dos enfermedades raras y sus diferencias


Referencias
- Grabowski GA, Petsko GA, Kolodny EH. Gaucher Disease. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; Accessed July 16, 2020.
- Grabowski GA, Kolodny EH, Weinreb NJ, Rosenbloom BE, Prakash-Cheng A, Kaplan P, Charrow J, Pastores GM, Mistry PK. Gaucher Disease: Phenotypic and Genetic Variation. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; Accessed July 16, 2020.
MAT-ES-2301496 V2 Septiembre 2025