- Artículo
- Fuente: Campus Sanofi
- 29 mar 2023
Diagnóstico en enfermedad de Pompe
Diagnóstico diferencial
La capacidad de diferenciar correctamente la enfermedad de Pompe de otras enfermedades es crucial para minimizar el retraso en el diagnóstico y optimizar los desenlaces de los pacientes.
Sin embargo, dado que la enfermedad de Pompe es rara y comparte muchos de sus signos y síntomas con otras enfermedades, el diagnóstico diferencial puede suponer un reto.
La enfermedad de Pompe debería valorarse cuando los signos y síntomas indiquen una degeneración muscular progresiva, en lactantes, niños adolescentes y adultos:
En lactantes: cardiomegalia /cardiomiopatía, hipotonía, rápida progresión de la debilidad muscular tanto del musculo ventilatorio como del esquelético, dificultades para alimentarse, y retraso del crecimiento.1,2,3,4
En niños, adolescentes y adultos: debilidad proximal de cinturas y/o de los músculos respitatorios además de escoliosis en adolescentes y elevación de los niveles de creatinquinasa (CK).5,6
Una vez surja la sospecha clínica, puede medirse la actividad de la enzima GAA para confirmar la enfermedad de Pompe.5,6
Considere la enfermedad de Pompe en el diagnóstico diferencial de estas otras enfermedades:1
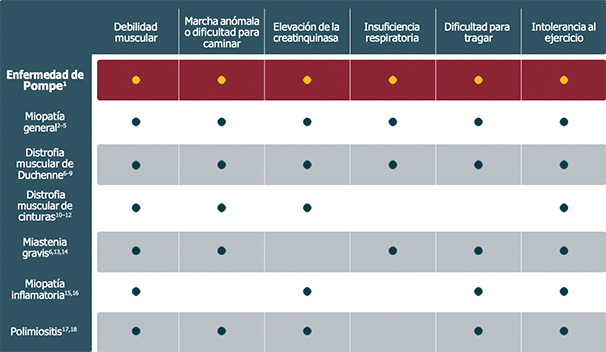
Poblaciones de alto riesgo
Como se muestra en las siguientes tablas, existen datos convincentes que respaldan la realización de pruebas rutinarias para detectar la enfermedad de Pompe (a partir de un análisis para detectar la enzima GAA) en poblaciones de pacientes que presentan ciertos síntomas sugestivos de la enfermedad de Pompe, tales como debilidad de cinturas y/o niveles elevados de creatinquinasa (CK), con o sin la debilidad de los músculos respiratorios:
Diagnósticos de la enfermedad de Pompe en pacientes con debilidad de cinturas.7,8,9,10
|
Autor |
Criterios para realizar pruebas |
Número de pacientes que se han sometido a pruebas |
Número de pacientes con enfermedad de Pompe |
|
oldstein (2009) |
Paciente adulto con debilidad muscular de la cintura o de las extremidades proximales y sospecha de enfermedad Pompe |
132 |
207132(15%) |
|
Preisler (2013) |
Distrofia muscular de la cintura y extremidades sin clasificar |
38 |
3/38 (8%) |
|
Willis (2012) |
Debilidad muscular de la cintura y extremidades ± síntomas respiratorios ± escoliosis ± columna vertebral rígida |
102 |
5/102 (4,9%) |
|
Bautista lorite (2013) |
Debilidad muscular de la cintura y extremidades |
144 |
15/144(10,2%) |
Diagnósticos de la enfermedad de Pompe en pacientes con creatina-cinasa (CK) elevada10,11,12
|
Autor |
Criterios para realizar pruebas |
Número de pacientes que se han sometido a pruebas |
Número de pacientes con enfermedad de Pompe |
|
Bautista lorite (2013) |
HiperCKmias idiopáticas |
205 |
5/205(2,4%) |
|
Fernandez (2006) |
HiperCKmias oligosintomática o asintomática idiopática |
104 |
4/104 (3,8%) |
|
Spada (2013) |
Pacientes con HiperCKemia asintomática idiopática |
137 |
3/137(2,2%) |
Pruebas diagnósticas y de confirmación
La enfermedad de Pompe es una enfermedad rara con signos y síntomas similares a los de otras muchas enfermedades.
Esto significa que la enfermedad de Pompe tiende a pasar desapercibida, al menos al principio, hasta que se han descartado otras enfermedades más frecuentes, lo que da como resultado un retraso en el diagnóstico que puede ser perjudicial o incluso potencialmente mortal.
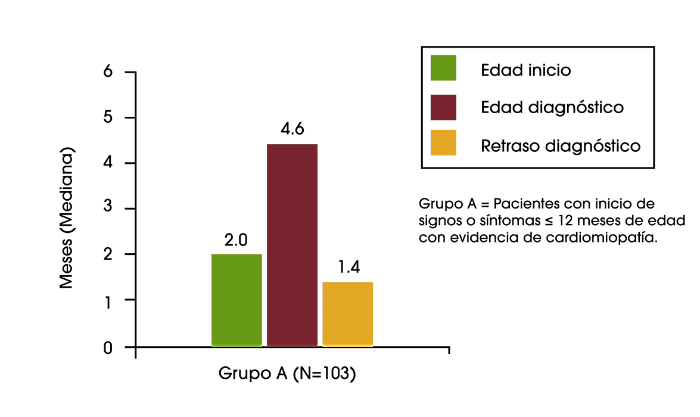
En lactantes, el diagnóstico temprano es especialmente importante, ya que la falta de tratamiento suele conllevar la muerte durante el primer año de vida. Sin embargo, se ha observado que los lactantes experimentan una mediana de retraso de 2,7 meses desde la primera aparición de los signos hasta el diagnóstico de la enfermedad de Pompe.1
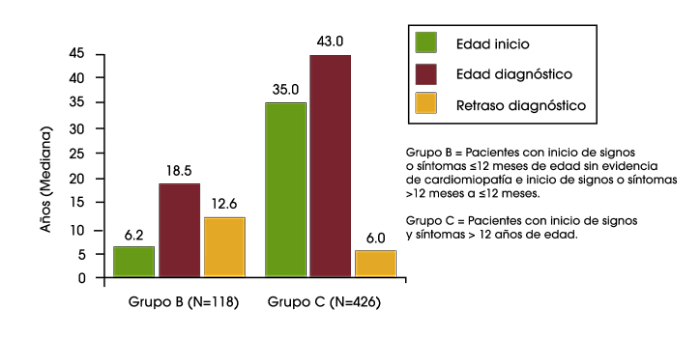
En un análisis reciente de los datos del Registro de Pompe se encontró retrasos diagnósticos en lactantes, niños y adolescentes/adultos. En los bebés que tuvieron el inicio de los síntomas durante los primeros 12 meses de vida, y que presentaban cardiomiopatía (es decir, la enfermedad de Pompe infantil clásica), el retraso diagnóstico medio fue de 1,4 meses. Para los pacientes con inicio de síntomas sobre la edad de 12 años, el retraso diagnóstico medio fue de 6,0 años. El retraso más largo en el diagnóstico fue una media de 12,6 años, que se encontró en los pacientes que tuvieron inicio de los síntomas durante los primeros 12 meses de vida, pero no tenían cardiomiopatía, o tuvieron el inicio de los síntomas entre los 12 meses y 12 años de edad.2
Por tanto, existe la necesidad de un diagnóstico precoz en todo el espectro de la enfermedad de Pompe, que ayude a optimizar los resultados del paciente.
Retraso del diagnósitco en lactantes, niños y adolescentes/ adultos con la enfermedad de Pompe2
La relación entre el genotipo y el fenotipo
La gravedad de la enfermedad y la edad de aparición están relacionadas con el grado de carencia enzimática;1 en general, el tipo de variante en GAA predice el fenotipo clínico;2 no obstante, hay factores como los genes modificadores y el ambiente, que pueden modular las relaciones entre el genotipo y el fenotipo, e influir en la edad de aparición, la gravedad y la evolución de la enfermedad de Pompe.3
Las variantes patogénicas pueden ser leves, polimórficas o graves (Figura 2).2 La herencia de dos variantes patogénicas graves suele traducirse en la ausencia total o casi total de actividad GAA y en la presencia de enfermedad de Pompe de inicio temprano. La herencia de una variante grave y otra leve, o de dos leves, suele traducirse en la presencia de actividad GAA residual y enfermedad de Pompe de inicio tardío.2 La presentación clínica y la evolución de la LOPD pueden variar incluso entre pacientes con el mismo genotipo, como pone de manifiesto la variabilidad intrafamiliar entre hermanos con LOPD.3
Diagnóstico
Aunque las vías clínicas para el diagnóstico de la enfermedad de Pompe son variables, el proceso implica generalmente:
Evaluación clínica de los síntomas presentes por un médico de atención primaria.
Remisión a un especialista para una investigación clínica en mayor profundidad, incluyendo pruebas analíticas o clínicas adicionales.
Pruebas de confirmación.
Diagnóstico definitivo
La enfermedad de Pompe se confirma por una ausencia completa o una disminución marcada de la actividad de la alfa glucosidasa ácida (GAA).3,4 La actividad residual de la GAA en pacientes con enfermedad de Pompe puede oscilar entre menos del 1 % (generalmente en lactantes) y el 40% de los niveles normales.5 La enfermedad de Pompe también puede confirmarse mediante el análisis de mutaciones, que muestra la presencia de dos alelos mutados.
Tradicionalmente, la prueba para detectar la enzima GAA se realizaba a partir de un cultivo de fibroblastos cutáneos.3,4 Sin embargo, la recogida de muestras es relativamente invasiva y transcurren aproximadamente 6 semanas hasta obtener los resultados. Se desaconseja claramente este prolongado período de espera, especialmente en lactantes con una enfermedad que progresa con rapidez. Por tanto, el uso de muestras de sangre, incluidas las muestras de sangre seca, se está convirtiendo en la práctica habitual. La extracción de muestras de sangre para la confirmación de la enfermedad de Pompe es mínimamente invasiva, exacta y puede generalmente ofrecer resultados en tan solo unos días.3,4
Si se encuentra una actividad enzimática reducida de GAA, se debe confirmar con una segunda muestra o por análisis genético.6
Aunque las biopsias musculares son una opción para las pruebas de actividad de la GAA, no suelen ser la opción preferida, ya que son invasivas y tienen un alto riesgo de falsos positivos si se manipulan las muestras de forma incorrecta. Las biopsias musculares pueden ser útiles para la evaluación histológica, pero es importante tener en cuenta que el contenido de glucógeno puede variar ampliamente entre los músculos, por lo que las biopsias aparentemente normales no descartan la enfermedad de Pompe.4 Así pues, un diagnóstico de la enfermedad de Pompe debe confirmarse siempre por la ausencia o reducción de la actividad de la GAA, o por un análisis genético.
¿Tienes alguna pregunta? Escríbenos para resolver tus dudas.
Consulta de Información Médica
Referencias
- Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8:267-88.
- Gilbert-Barness E. Review: Metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 2004; 34:15-34.
- Howell RR, Byrne B, Darras BT, Kishnani P, Nicolino M, van der Ploeg A. Diagnostic challenges for Pompe disease: An under-recognized cause of floppy baby syndrome. Genet Med 2006:8;1-8.
- Gilchrist JM. Overview of neuromuscular disorders affecting respiratory function. Semin Respir Crit Care Med 2002; 23:191-200.
- American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009;40:149-60.
- Ausems MG, Lochman P, van Diggelen OP, Ploos van Amstel HK, Reuser AJ, Wokke JH. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology. 1999 Mar 10;52(4):851-3.
- Goldstein JL, Young SP, Changela M, Dickerson GH, Zhang H, Dai J, Peterson D, Millington DS, Kishnani PS, Bali DS (2009). Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve 40:32-36.
- Preisler N, Lukacs Z, Vinge L, Madsen KL, Husu E, Hansen RS, Duno M, Andersen H, Laub M, Vissing J (2013). Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol Genet Metab 110(3):287-9.
- Willis T, Roberts M, Hilton-Jones D, Quinlivan R,5 Hanna M, Straub V (2012). Detection rate of Pompe disease in undiagnosed neuromuscular patients from four major centre’s in the UK- Results of a 12 month prospective audit. BMC Musculoskelet Disord 14(Suppl 2):P20.
- Bautista Lorite J (2013) Detección de la enfermedad de Pompe en pacientes con distrofia de cinturas indefinidas o hiperCKemias asintomáticas. Expert Rev Neur Ed especial Octubre 2013:17-19.
- Fernandez C, de Paula AM, Figarella-Branger D, Krahn M, Giorgi R, Chabrol B, et al. (2006). Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 66:1585-7.
- Spada M, Porta F, Vercelli L, Pagliardini V, Chiadò-Piat L, Boffi P, Pagliardini S, Remiche G, Ronchi D, Comi G, Mongini T (2013). Screening for later-onset Pompe's disease in patients with paucisymptomatic hyperCKemia. Mol Genet Metab 109:171-173.
- Kishnani PS, Hwu W-L, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006; 148:671-676.
- Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J; Pompe Registry Boards of Advisors. Timing of diagnosis of patients with Pompe disease: data from the Pompe registry. Am J Med Genet A. 2013;161A(10):2431-43.
- Zhang H, Kallwass H, Young SP, et al. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet med 2006; 8:302-306.
- Winchester B, Bali D, Bodamer OA, et al for The Pompe Disease Diagnostic Working Group. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008;93(3):275-281.
- Van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372(9646):1342-53
- American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009;40:149-60.
MAT-ES-2301236 V2 Septiembre 2025