- Artículo
- Fuente: Campus Sanofi
- 10 abr 2024
¿Cómo identificar a pacientes que pueden apuntar a una MPS I?
¿Qué es la mucopolisacaridosis tipo I (MPS I)?
La MPS I es una enfermedad autosómica recesiva infradiagnosticada que puede variar entre las formas más graves a atenuadas; pero todos los pacientes con MPS I se enfrentan a manifestaciones progresivas y potencialmente mortales.1–3
La MPS I aparece por variantes patogénicas del gen de la α-L-iduronidasa (IDUA) que deriva
en una deficiencia en la actividad de la enzima IDUA.1,2
Esta actividad deficiente de la enzima IDUA inicia una cascada de acumulación de glicosaminoglicanos lisosomales (GAG), daño progresivo de tejidos y órganos, pérdida funcional, deterioro clínico y discapacidad progresiva.1-2
Aclaramiento de GAG en personas sanas sin MPS I1
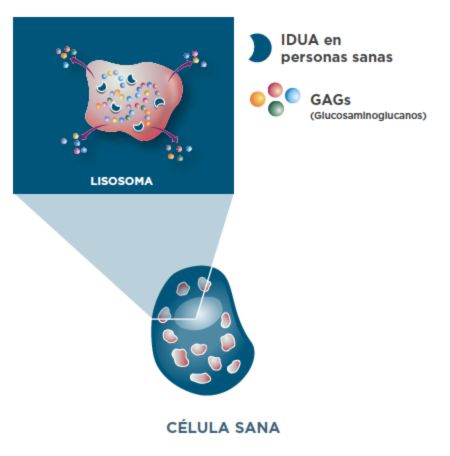
La enzima IDUA metaboliza dos tipos de GAG: dermatán sulfato y heparán sulfato.1
Acumulación de GAG en pacientes con MPS I1
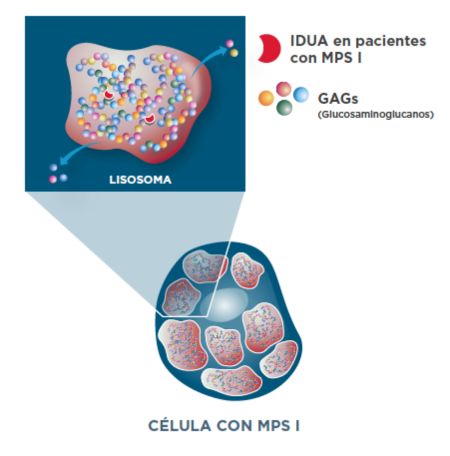
Las variantes patogénicas en el gen IDUA llevan a una actividad enzimática IDUA deficiente, lo que resulta en la acumulación lisosomal de GAG en muchos tipos de células y tejidos.1
El espectro de la enfermedad MPS I está marcado por su heterogeneidad, y puede complicar su diagnóstico, por ejemplo, la edad de aparición es variable, presenta una combinación atípica de síntomas y una amplia variedad de manifestaciones clínicas irreversibles y potencialmente mortales.1–3
El reconocimiento y diagnóstico temprano de los síntomas es fundamental para alcanzar el mejor pronóstico a largo plazo en pacientes con MPS I.1
Piense más allá de lo habitual cuando los pacientes presenten una gran variedad de síntomas
Debido a la amplia variabilidad en la presentación clínica, la progresión moderada y el solapamiento con otras enfermedades, los pacientes con MPS I atenuada pueden pasar años antes de ser diagnosticados o de recibir un tratamiento adecuado.1,5
La naturaleza progresiva de la MPS I hace que un retraso en el tratamiento pueda tener un impacto significativo en el pronóstico a largo plazo de los pacientes con MPS I.1,6
Es fundamental que distintos especialistas estén familiarizados con los síntomas de la MPS I para facilitar un diagnóstico más temprano y el inicio de un tratamiento efectivo.1,6
Retrasos en el diagnóstico y tratamiento de pacientes con MPS I atenuada1 | |||
|
2–4 años |
Retraso medio entre la presentación de los síntomas y el diagnóstico1 |
4–8 años |
Retraso medio entre el diagnóstico y el inicio del tratamiento específico de la enfermedad1 |
Signos y síntomas que se observan en ≥25% de los diagnósticos confirmados de MPS I7
|
Rasgos faciales toscos |
Deterioro cognitivo |
|
Valvulopatía |
Macroglosia |
|
Opacidad corneal |
Esplenomegalia |
|
Hepatomegalia |
Obstrucción de la trompa de Eustaquio |
|
Obstrucción de las vías respiratorias superiores |
Otitis media |
|
Apnea obstructiva del sueño |
Displasia de cadera |
|
Cifosis |
Genu valgo |
|
Contracturas articulares |
Enfermedad reactiva de las vías respiratorias |
|
Hernia |
Escoliosis |
|
Disostosis múltiple |
Síndrome del túnel carpiano |
Si sospecha la presencia de MPS I: realice una prueba o derive al paciente para que reciba un diagnóstico definitivo.
Cribado y diagnóstico de la MPS I
Cribado de primera línea7
Ensayo de actividad enzimática IDUA utilizando una muestra de gota de sangre seca (DBS): Puede corroborar la sospecha de MPS I, pero se deben confirmar los resultados con pruebas de diagnóstico adicionales.
El diagnóstico precoz y el inicio de un tratamiento específico de la enfermedad pueden cambiar el curso de la MPS I.1,7
Si sospecha la presencia de MPS I, realice una prueba o derive al paciente para que reciba un diagnóstico definitivo.
Diagnóstico definitivo7

Pruebas bioquímicas: Actividad de la enzima IDUA en leucocitos sanguíneos, suero o plasma.
Pruebas genéticas: Pruebas de genética molecular para confirmar variantes patogénicas del gen IDUA.

DBS: gota de sangre seca
GAG: glicosaminoglicano
IDUA: α-L-iduronidasa
MPS I: mucopolisacaridosis tipo I DE, desviación estándar
- Beck M et al. Genet Med 2014; 16(10): 759–765.
- de Ru M et al. Orphanet J Rare Dis 2011; 6: 55.
- Moore D et al. Orphanet J Rare Dis 2008; 3 (3)3: 24.
- Martins A et al. BMC Endocr Disord 2018; 8(1): 83.
- D’Aco K et al. Eur J Pediatr 2012; 171(6): 911–919.
- Lehman T et al. Rheumatology 2011; 50: 41–48.
- Clarke L et al. J Pediatr 2017; 182: 363–370.
MAT-ES-2302781 V1 Enero 2024