- Artículo
- Fuente: Campus Sanofi
- 17 abr 2023
Diagnóstico y pruebas de la MPS I

Detección temprana
La detección temprana de signos y síntomas de la MPS I puede evitar el deterioro multiorgánico de los pacientes con MPS I, que podría llegar a ser irreversible y mortal1-3

Fenotipos
La MPS I muestra diversos fenotipos, de grave a atenueado, con diferentes manifestaciones multiorgánicas, algunas de ellas progresivas y potencialmente mortales1,4,5

Retraso en el diagnóstico
La variabilidad clínica de la MPS I puede retrasar e diagnóstico y el tratamiento, afectando a la salud y al bienestar del paciente a lo largo de la vida1,2

Muestra de sangre en gota seca
El análisis en muestra de sangre en gota seca es una prueba sencilla y fiable que resulta útil para el diagnóstico de la MPS I3,6

Prueba de hermanos
Someter a los hermanos y hermanas de los pacientes con MPS I a las pruebas facilita el diagnóstico temprano y el tratamiento específico a largo plazo3
Si sospechas
Pídenos gratuitamente el kit de diagnóstico del test DBS. Te lo enviamos a tu consulta.
Identificarla a tiempo puede cambiar su evolución7
La aparición de la enfermedad suele producirse en la edad pediátrica, pero los signos son leves y pueden ignorarse o diagnosticarse erróneamente.
Un cuadro clínico frecuente es el del paciente con aparición de contracturas articulares no inflamatorias en la edad pediátrica, diagnosticadas inicialmente como artritis reumatoide, artritis idiopática juvenil o esclerodermia y que, tras muchos años (hasta 50 años) de diagnóstico erróneo, se identifican como signos de MPS.
La forma atenuada de MPS puede permanecer aparentemente asintomática durante años, con una progresión silente de la enfermedad y no reconocerse hasta un estado avanzado, cuando es demasiado tarde para poder intervenir.
Se debe ampliar el conocimiento de la MPS por parte de los médicos especialistas para que puedan desempeñar un papel importante, no solo en el tratamiento, sino también en el diagnóstico de la MPS.
Detéctalo a tiempo y podrá marcar la diferencia8
El retraso del diagnóstico es común en pacientes con MPS I atenuada, los cuales sufren durante años, a veces décadas, esta enfermedad progresiva y debilitante antes de que sea finalmente reconocida.6,9,10
Un diagnóstico precoz y un inicio temprano de la terapia enzimática de sustitución cambian sustancialmente la historia natural de la forma atenuada de mucopolisacaridosis de tipo I.8
Diagnóstico diferencial
Es fundamental distinguir correctamente la MPS I de otros trastornos para reducir al mínimo el retraso del diagnóstico y optimizar los resultados del tratamiento.
Sin embargo, la MPS I puede ser muy difícil de detectar debido a su rareza, su presentación variable, así como la similitud de los síntomas clínicos con los de otras enfermedades.11 El retraso en el diagnóstico es máximo en las personas que presentan la forma atenuada de MPS I.12 Debido a su afectación osteomuscular característica, la MPS I atenuada suele confundirse con enfermedades reumáticas.11,13,14
- Otros tipos de mucopolisacaridosis
- Artritis reumatoide (juvenil)
- Artrogriposis
- Enfermedades del tejido conjuntivo
- Polimiositis
- Síndrome del túnel carpiano
Debe sospecharse MPS I en personas que presenten uno o más de los siguientes signos:
✓ Cifosis/deformidad en giba
✓ Tosquedad facial
✓ Opacidad corneal
✓ Deterioro cognitivo/del desarrollo psicomotor
✓ Hepatoesplenomegalia
✓ Contracturas articulares no inflamatorias
✓ Alteraciones de las válvulas cardíacas (indicativos de MPS I grave).11-13,15,16
✓ Hernias recurrentes
✓ Opacidad corneal
✓ Síndrome del túnel carpiano o tosquedad facial (indicativos de MPS I atenuada).11,12
¿Sospecha de MPS I?12,13
- El diagnóstico es fácil de realizar.
- Un sencillo análisis de orina puede detectar la presencia de GAGs en pacientes con sospecha.
- El diagnóstico de MPS I se basa en la detección de una disminución de la actividad enzimática de α-L-Iduronidasa (IDUA).

Pruebas diagnósticas y confirmatorias
El diagnóstico de la MPS1 es sencillo y rápido, y se basa en la demostración de una deficiencia profunda de la enzima α-L-iduronidasa1,17.
En pacientes con signos y síntomas indicativos de MPS I, el análisis de GAG en orina suele ser la primera prueba diagnóstica, aunque el diagnóstico se confirma midiendo la actividad enzimática.

Exámenes urinarios
Análisis cuantitativo: elevación de los glicosaminoglicanos**.
Análisis cualitativo: demostración de fracciones anormales de dermatán sulfato y heparán sulfato.
En las formas leves, los niveles urinarios de GAG** pueden ser normales o estar ligeramente aumentados
Si la orina está demasiado diluida (densidad <1,015) se pueden obtener resultados falsos negativos.9

Análisis de sangre
El diagnóstico definitivo se establece mediante la determinación de la actividad de la α-L-iduronidasa en leucocitos, en suero o en una gota de sangre seca (DBS, por sus siglas en inglés).18 La deficiencia de α-L-iduronidasa confirma el diagnóstico de MPS I. Si el análisis es negativo, deben realizarse otros análisis enzimáticos para confirmar o descartar otro tipo de MPS.
El valor de la actividad enzimática no permite distinguir una forma grave de una forma atenuada.
La búsqueda de mutaciones en el gen IDUA que codifica la α-L-iduronidasa (4p16.3) puede, para ciertas mutaciones, permitir predecir la gravedad del fenotipo17.
* trasplante de células madre hematopoyéticas
** glicosaminoglicanos
Estudio de los hermanos
Si una persona padece MPS I, es muy probable que otros miembros de su familia también tengan el gen defectuoso. Es muy importante estudiar a los hermanos de un paciente con MPS I para determinar si también están afectados. La prueba necesaria para ello se denomina «cribado de familiares» o «segregación familiar».
Cribado neonatal
Se han desarrollado varias técnicas de cribado con gotas de sangre seca y un análisis múltiple de alto rendimiento que permite realizar un cribado neonatal para la detección selectiva de un grupo de enfermedades metabólicas, entre ellas la MPS I.19 En España actualmente no está incluido este cribado en ninguna comunidad autónoma.
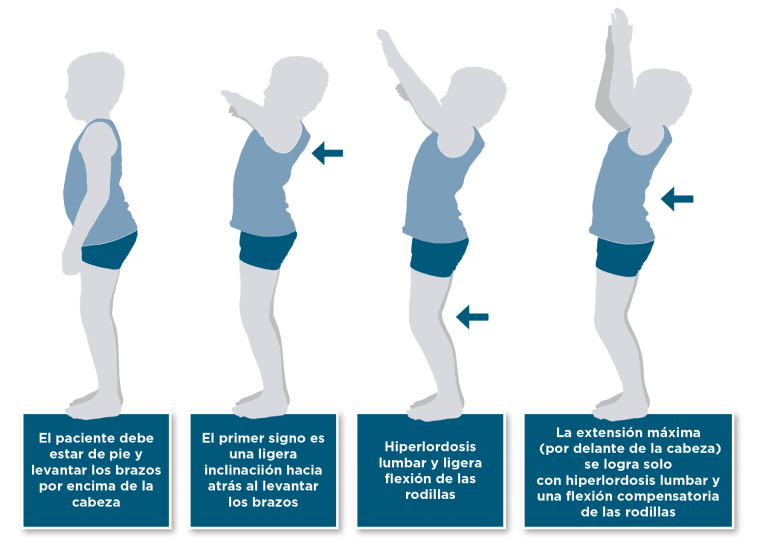
Prueba de rango de movilidad de hombros
La prueba de rango de movilidad de hombros facilita el diagnóstico precoz de la MPS20
Es frecuente que los pacientes con mucopolisacaridosis (MPS) no sean capaces de estirar los brazos por encima de la cabeza. Por eso, se debe prestar atención a los siguientes movimientos y a la posición final. Si además no hay signos inflamatorios en el diagnóstico analítico, debe considerarse un claro indicio de la presencia de MPS y debería confirmarse mediante una muestra de sangre en gota seca.21
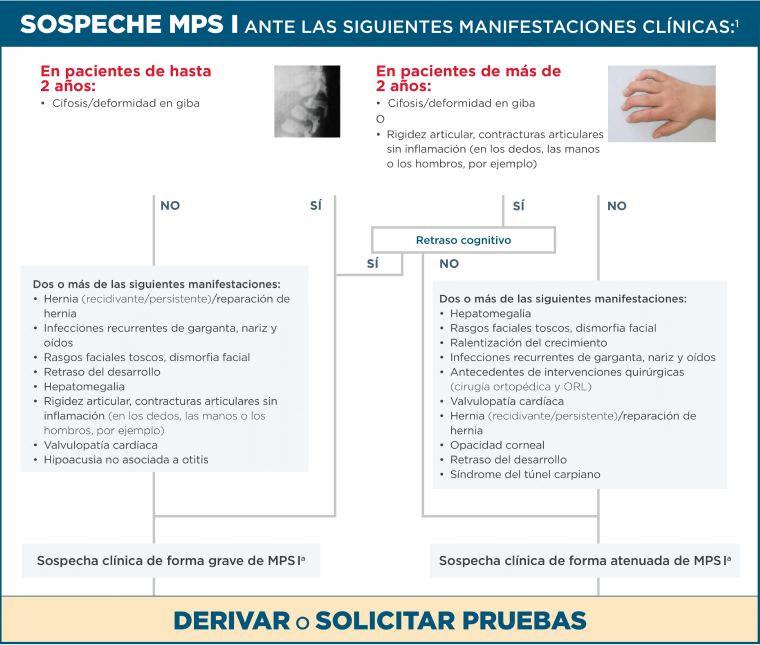
Algoritmo diagnóstico
Los pacientes con MPS I presentan combinaciones de síntomas sin relación aparente
Ante la presencia de cifosis, contracturas articulares o ambas, acompañadas de otros síntomas menos específicos, se debería derivar al paciente o solicitar las pruebas diagnósticas de la MPS I22

Referencias
GAGs: glucosaminoglucanos; MPS I: mucopolisacaridosis de tipo I; ORL: otorrinolaringología.
- Beck M, et al. Genet Med. 2014;16(10):759-765.
- D’Aco K, et al. Eur J Pediatr. 2012;171(6):911-919.
- Lehman TJ, et al. Rheumatology. 2011;50(suppl 5):v41-v48.
- Giugliani R, et al. Genet Mol Biol. 2010;33(4):589-604.
- Soni-Jaiswal A, et al. Orphanet J Rare Dis. 2016;11(1):96.
- Cimaz R, et al. Pediatr Rheumatol Online J. 2009;7:18.
- Rigoldi M, Verrecchia E, Manna R, et al. Clinical hints to diagnosis of attenuated forms of Mucopolysaccharidoses. Italian Journal of Pediatrics 2018;44(Suppl 2):132.
- Gabrielli O, Clarke LA, Ficcadenti A, et al. 12 year follow up of enzyme-replacement therapy in two siblings with attenuated mucopolysaccharidosis I: the important role of early treatment. BMC Medical Genetics 2016;17:19.
- Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29.
- Vijay S, Wraith JE. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr. 2005;94(7):872-877.
- Murphy AM, Lambert DM, Treacy EP, O’Meara A, Lynch SA. Incidence and prevalence of mucopolysaccharidosis type 1 in the Irish Republic. Arch Dis Child 2009;94:52-4.
- Neufeld EF and Muenzer J. (2001) The mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, and Vogelstein B. (eds.). 8th edition, Vol. III. McGraw-Hill, Medical Publishing Division, pp. 3421
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249-54.
- Moore D, et al. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis 2008;3:24.
- Lin HY, Lin SP, Chuang CK, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984-2004. Am J Med Genet A 2009;149A:960-4.
- Malm G, Lund AM, Mansson JE, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta Paediatr 2008;97:1577-81.
- Protocole National de Diagnostic et de Soins MPS – HAS – Juillet 2016
- Lehman TJA, Miller N, Norquist B et al. Diagnosis of the mucopolysaccharidoses. Rheumatology 2011;50:v41-v18
- Johnson BA, Dajnoki A, Bodamer OA. Diagnosing lysosomal storage disorders: mucopolysaccharidosis type I. Curr Protoc Hum Genet. 2015 Jan 20;84:17.17.1-8.
- Foster HE, Jandial S. pGALS - paediatric Gait Arms Legs and Spine: a simple examination of the musculoskeletal system. Pediatr Rheumatol Online J 2013;11(1):44
- Guffon N, et al. Growth impairment and limited range of joint motion in children should raise suspicion of an attenuated form of mucopolysaccharidosis: expert opinion. Eur J Pediatr 2019;178(4):593–603.
- Tylki-Szymańska A, et al. Acta Paediatr. 2018;107(8):1402-1408.
MAT-ES-2503840 V1 Diciembre 2025