- Artículo
- Fuente: Campus Sanofi
- 26 mar 2024
Fisiopatología MPS I

La MPS I es uno de los más de cincuenta trastornos hereditarios raros que reciben el nombre de enfermedades de depósito lisosomal. Cada una de estas enfermedades está causada por un defecto genético congénito que provoca la deficiencia de una o varias enzimas lisosómicas concretas. La edad de inicio, los órganos y sistemas afectados, así como la intensidad de estos trastornos varían notablemente entre sí, pero todos son progresivos.
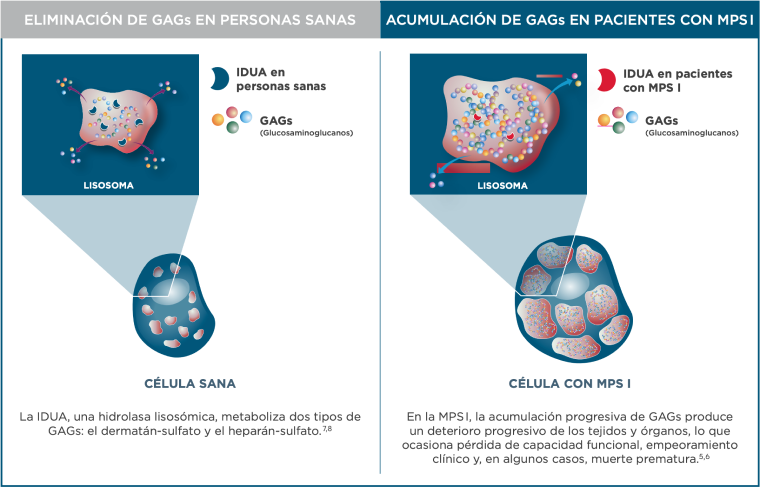
La MPS I es una enfermedad hereditaria autosómica recesiva que cursa con manifestaciones anatomopatológicas en la mayoría de los sistemas y tejidos del organismo1. La enfermedad está causada por un defecto en el gen que codifica la enzima lisosómica α-L-iduronidasa que hace que las personas afectadas sean incapaces de producir la enzima o la produzcan en cantidades bajas. La consiguiente deficiencia de α-L-iduronidasa altera la capacidad de las células para degradar los glucosaminoglucanos (GAG) dermatán y heparán sulfato, lo que provoca la acumulación constante de GAG en las células1. Esto desencadena una lenta cascada de procesos patológicos específicos en los órganos y la pérdida de su función2. A medida que progresa la enfermedad, la mayor parte del daño celular y tisular se vuelve irreversible.
Al nacer, los bebés afectados parecen inicialmente normales desde el punto de vista clínico, pero con el tiempo, la acumulación progresiva de GAG provoca un deterioro de las funciones motriz, respiratoria y cardíaca y un aumento del tamaño de algunos órganos, como el hígado y el bazo1,3. Aunque presenten un grado semejante de deficiencia enzimática, los pacientes con MPS I pueden experimentar una amplia variedad de síntomas de grado muy variable1,4.
La MPS I se produce por mutaciones en el gen de la a-L-iduronidasa (IDUA), que llevan a un déficit de actividad IDUA, y a la acumulación progresiva de GAGs en numerosos tipos de células y tejidos5,6


¿Tienes alguna pregunta? Escríbenos para resolver tus dudas.
Consulta de Información Médica
Referencias
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001:3421-52.
- Clarke LA. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev Mol Med. 2008;10:e1.
- Arn P, Wraith J, Underhill L. Characterization of surgical procedures in patients with mucopolysaccharidosis type I: findings from the MPS I Registry. J Pediatr. 2009; 154:859-64 e3.
- Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123:19-29.
- Beck M, et al. Genet Med. 2014;16(10):759-765.
- Giugliani R, et al. Genet Mol Biol. 2010;33(4):589-604.
- Neufeld EF, et al. The mucopolysaccharidoses. In: Scriver CR, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 1995:2465-2494.
- Scott, et al. Am J Hum Genet. 1990:47(5)_802-207.
- Lehman TJ, et al. Rheumatology. 2011;50(suppl 5):v41-v48.
- Pastores GM, et al. Mol Genet Metab. 2007;91(1):37-47.
MAT-ES-2503840 V1 Diciembre 2025