- Artículo
- Fuente: Campus Sanofi
- 26 mar 2024
Progresión y pronóstico de la enfermedad MPS I

Progresión y pronóstico
Debido a la acumulación crónica y progresiva de glucosaminoglucanos en los lisosomas de todas las células del organismo, los pacientes afectados por la MPS I presentan una disfunción multiorgánica que conlleva una morbilidad considerable y una mortalidad prematura en los más gravemente afectados. Al igual que la mayoría de las demás enfermedades metabólicas, la MPS I es muy heterogénea, de modo que la edad de comienzo, los órganos y sistemas afectados, la gravedad y la velocidad de progresión de la enfermedad varían considerablemente.1
En los lactantes y niños pequeños gravemente afectados, los síntomas aparecen poco después del nacimiento y progresan rápidamente.1 Sin tratamiento, aproximadamente el 75% de estos niños fallecen antes de cumplir 10 años,2 habitualmente por enfermedad obstructiva de las vías respiratorias, infecciones respiratorias y complicaciones cardíacas.3
En niños y adultos con formas atenuadas de MPS I, la evolución de la enfermedad es mucho más variable. Las manifestaciones aparecen al principio de la infancia, progresan más despacio y son menos evidentes que en los pacientes gravemente afectados. Los pacientes con MPS I atenuada pueden vivir hasta la edad adulta, pero con una morbilidad importante y una disminución en la esperanza de vida.3
Reconocer las manifestaciones tempranas más frecuentes de los diferentes fenotipos de la MPS I es clave para su diagnóstico
Los pacientes con MPS I presentan signos y síntomas muy diversos1
La variabilidad clínica de la MPS I puede retrasar el diagnóstico y el tratamiento, afectando a la salud y al bienestar del paciente a lo largo de su vida3,4
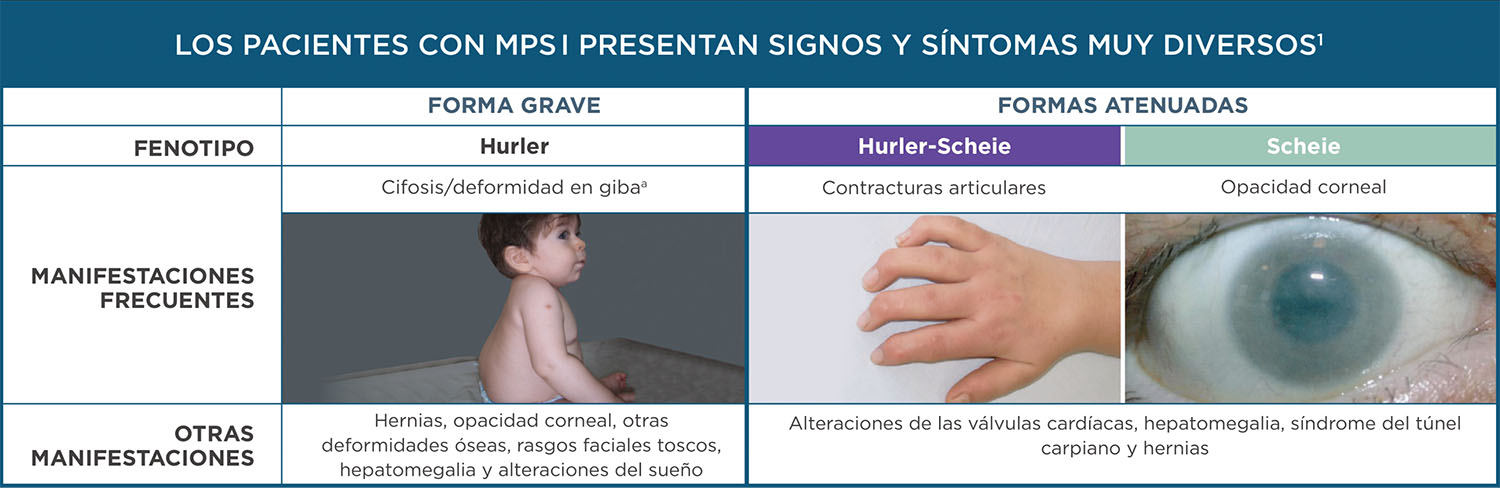
a. Fotografía por cortesía del Dr. Guelbert
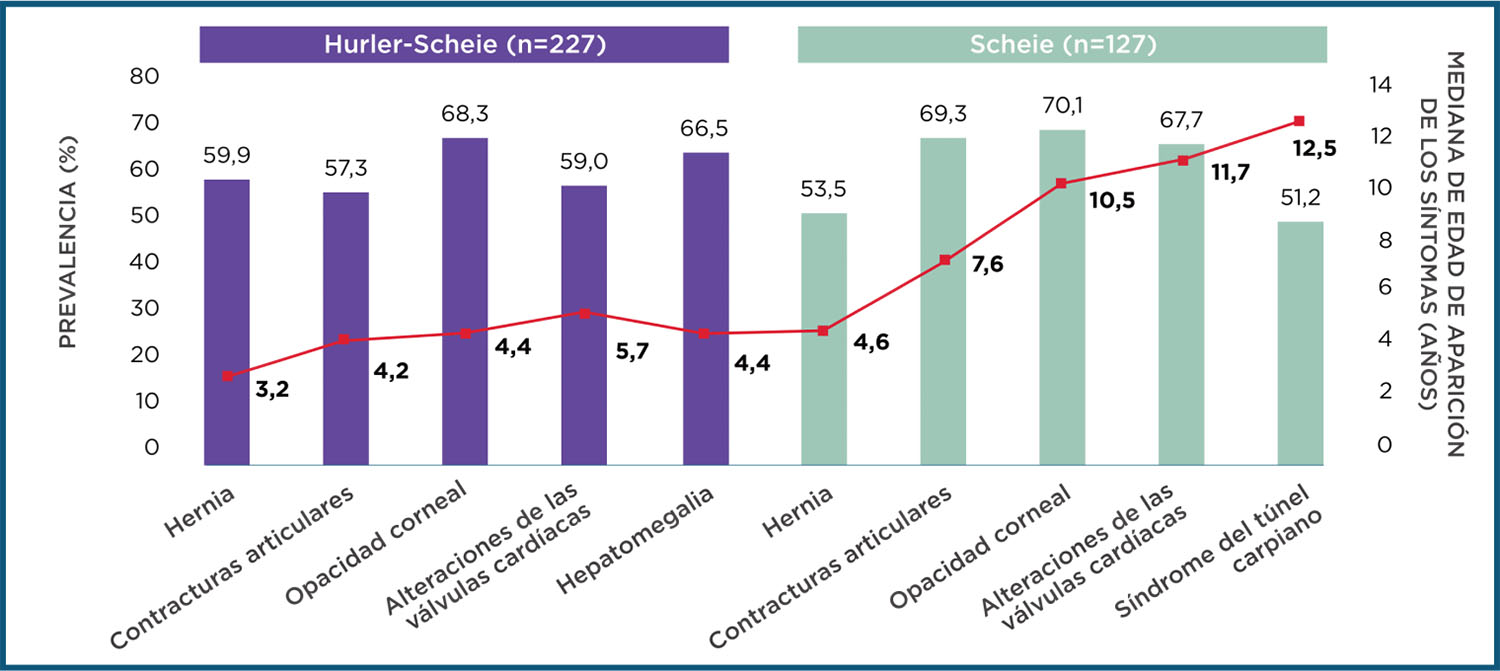
Las formas atenuadas de la MPS I
Afectan a distintos órganos y sistemas. La edad de aparición de los síntomas es muy variable
La acumulación de GAG en las células
Comienza al principio de la vida y progresa hasta provocar complicaciones debilitantes
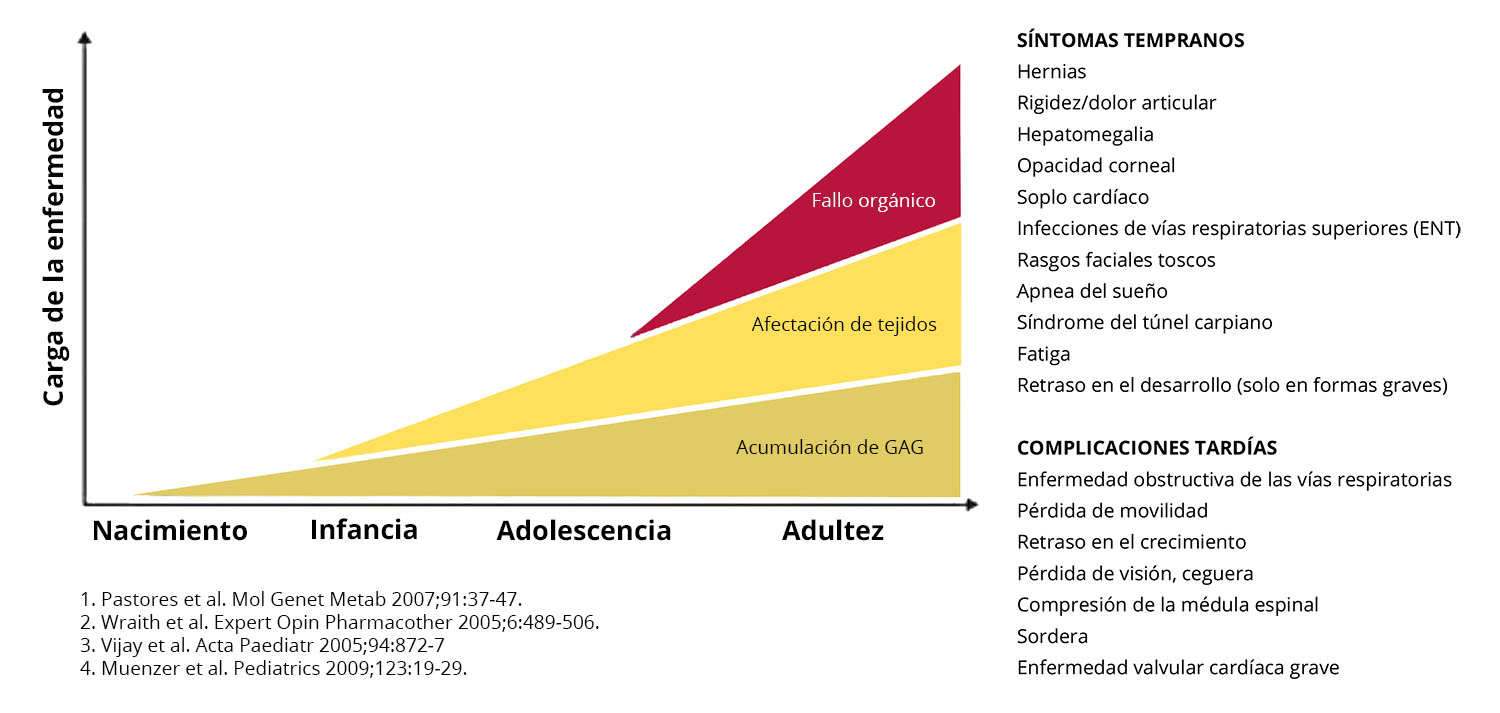
Síntomas Iniciales
- Hernias
- Rigidez/dolor articular
- Hepatomegalia
- Opacidad corneal
- Soplo cardíaco
- Infecciones ORL
- Facies tosca
- Apneas del sueño
- Síndrome del túnel carpiano
- Astenia
- Retraso desarrollo (solo en la forma grave)
Complicaciones tardías
- Enfermedad obstructiva de las vías respiratorias
- Pérdida de movilidad
- Retraso del crecimiento
- Pérdida de visión, ceguera
- Compresión medular
- Sordera
- Valvulopatía cardíaca grave

¿Tienes alguna pregunta? Escríbenos para resolver tus dudas.
Consulta de Información Médica
Referencias
MPS I: mucopolisacaridosis de tipo I.
- Beck M et al. Genet Med. 2014;16(10):759-765.
- Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis 2008;3:24.
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001:3421-52.
- Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123:19-29.
MAT-ES-2503840 V1 Diciembre 2025