- Artículo
- Fuente: Campus Sanofi
- 20 oct 2024
¿Qué es la enfermedad de Pompe?
¿Qué es la enfermedad de Pompe?
La enfermedad de Pompe, también conocida como glucogenosis tipo II o déficit de maltasa ácida, es una enfermedad con herencia autosómica recesiva que se produce por un déficit de la enzima alfa-glucosidasa ácida.
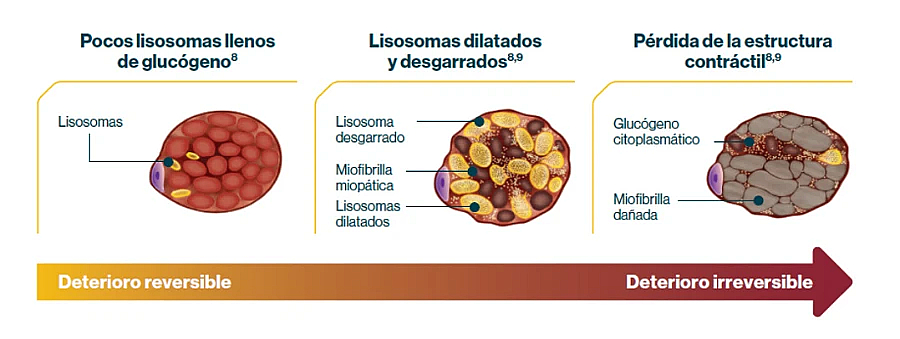
La alfa-glucosidasa ácida es fundamental para la degradación del glucógeno en el interior de los lisosomas de las células humanas de todos los tejidos. Cuando existe un déficit de esta enzima, el glucógeno se acumula en el interior de los lisosomas sin degradarse. Las células se van llenando de forma progresiva de lisosomas cargados con glucógeno que impiden su normal funcionamiento, bloqueando de este modo el proceso autofágico de las células. Consecuentemente, el citoplasma celular se llena de un material de desecho celular que no es procesado de forma correcta, y finalmente, existe una disfunción celular que lleva a la muerte de las células afectadas. No se conoce bien el motivo por el que los tejidos que más sufren esta alteración son el músculo esquelético, liso y cardíaco, y las neuronas del sistema nervioso central. En el caso del músculo esquelético, las fibras musculares se llenan de lisosomas repletos de glucógeno y de vacuolas autofágicas, lo que altera el funcionamiento del aparato contráctil de la fibra muscular. En fases avanzadas, los lisosomas se rompen, llenando el sarcoplasma de glucógeno libre que lleva a una disfunción de la fibra muscular y a su muerte.
La acumulación de glucógeno lisosómico conduce al deterioro progresivo e irreversible del músculo5,7
La actividad enzimática residual determina el momento de inicio de la enfermedad. Así, los casos infantiles se producen en pacientes con una actividad ausente o menor al 1%, mientras que los casos de inicio tardío, juveniles o del adulto, se producen en pacientes con una actividad de entre el 2% y el 30%.
1. Kishnani PS, et al. Genet Med. 2006;8(5):267-288. 2. Kishnani PS, et al. J Pediatr. 2004;144(5 suppl):S35-S4. 3.American Association of Neuromuscular and Electrodiagnostic Medicine. Muscle Nerve. 2009;40(1):149-160. 4. Hirschhorn R, et al. In: Scriver CR, et al. eds. The Metabolic Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3389-3420. 5.Cupler EJ, et al. Muscle Nerve. 2012;45(3):319-333. 6. Chien YH, et al. J Pediatr. 2011;158(6):1024-1027.e1. 7. Al Jasmi F, et al. BMC Neurology. 2015;15:205. 8. Thurberg BL,et al. Lab Invest. 2006;86(12):1208-1220. 9. Raben N, et al. Mol Genet Metab. 2010;101(4):324-331.
Incidencia y prevalencia
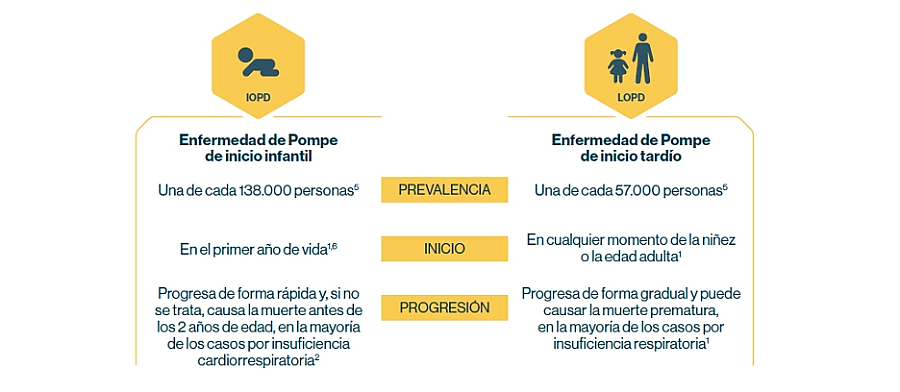
La enfermedad de Pompe es heterogénea: la edad de aparición y el ritmo de progresión son variables.
Como ocurre con todas las enfermedades raras, es difícil saber con exactitud cuántas personas están afectadas en realidad. Extrapolando las supuestas cifras de incidencia, se estima que la prevalencia mundial en la actualidad puede ser de 5000 a 10 000 personas (de ambos sexos y de edades y etnias diferentes)1.
IOPD: enfermedad de Pompe de inicio infantil; LOPD: enfermedad de Pompe de inicio tardío
1. Kishnani PS, et al. Genet Med. 2006;8(5):267-288. 2. Kishnani PS, et al. J Pediatr. 2004;144(5 suppl):S35-S43. 3. American Association of Neuromuscular and Electrodiagnostic Medicine. Muscle Nerve. 2009;40(1):149-160. 4. Chien YH, et al. J. 3. Pediatr. 2011;158(6):1024-1027.e1. 5. Ausems MGEM, et al. Eur J Hum Genet. 1999;7(6):713-716. 6. Kishnani PS, et al. J Pediatr. 2006;148(5):671-676. 7. Alejaldre A, et al. Neuromuscul Disord. 2012;22(suppl 2):S148-S154.
Distribución étnica
Varios estudios indican que la incidencia de la enfermedad de Pompe puede variar entre las poblaciones, oscilando entre 1 caso por cada 14 000 y 1 caso por cada 300 000, en función de la zona geográfica o del grupo étnico examinado.13 En lactantes, la enfermedad de Pompe parece ser más frecuente13:
-
Entre los afroamericanos
-
En el sur de China y en Taiwán
En cuanto a los adultos con enfermedad de Pompe, hay una incidencia comparativamente alta de la enfermedad en los Países Bajos.10 Además, se ha descubierto que algunas mutaciones específicas del gen GAA son más frecuentes en ciertos grupos (ver la sección “Mutaciones” que aparece a continuación).
1. Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counseling. Eur J Hum Genet 1999 Sep; 7(6): 713-6. 2. Hirschhorn R, Reuser AJ. Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G., eds. The Online Metabolic and Molecular Bases of Inherited Disease. OMMBID.
Genética de la enfermedad de Pompe
La enfermedad de Pompe se produce por un déficit de la enzima lisosómica α-glucosidasa ácida (GAA) y se hereda de forma autosómica recesiva.1 La localización de la mutación en el gen GAA y el tipo de mutación suelen predecir el fenotipo clínico de la enfermedad de Pompe.2
Herencia autosómica recesiva ligada al cromosoma 17 y frecuencia alélica
Hasta la fecha se han descrito más de 300 mutaciones del gen GAA, localizado en el cromosoma 17.1,3 La enfermedad de Pompe se hereda de forma autosómica recesiva. En el caso más habitual, ambos progenitores son portadores asintomáticos.1
En la Figura 1 se muestra el patrón de herencia autosómica recesiva con dos progenitores portadores.1
Probabilidades en cada embarazo:1
|
25% |
25% |
50% |
|
El hijo recibe dos alelos defectuosos o patogénicos y hereda la enfermedad de Pompe |
El hijo recibe dos alelos normales y no se ve afectado |
El hijo recibe un alelo normal y uno defectuoso, y se convierte en portador |
Cabe señalar que hay nuevos datos de familias con varias generaciones de individuos afectados por las formas temprana y tardía de la enfermedad de Pompe (IOPD y LOPD, respectivamente).3 Parece que la frecuencia alélica de las mutaciones del gen GAA es mucho mayor de lo que se pensaba y que puede haber distintas variantes genéticas en una misma familia. La posibilidad de que los familiares cercanos de un paciente con LOPD o IOPD tengan otros parientes afectados exige ampliar el asesoramiento genético y las pruebas de detección a los familiares.3
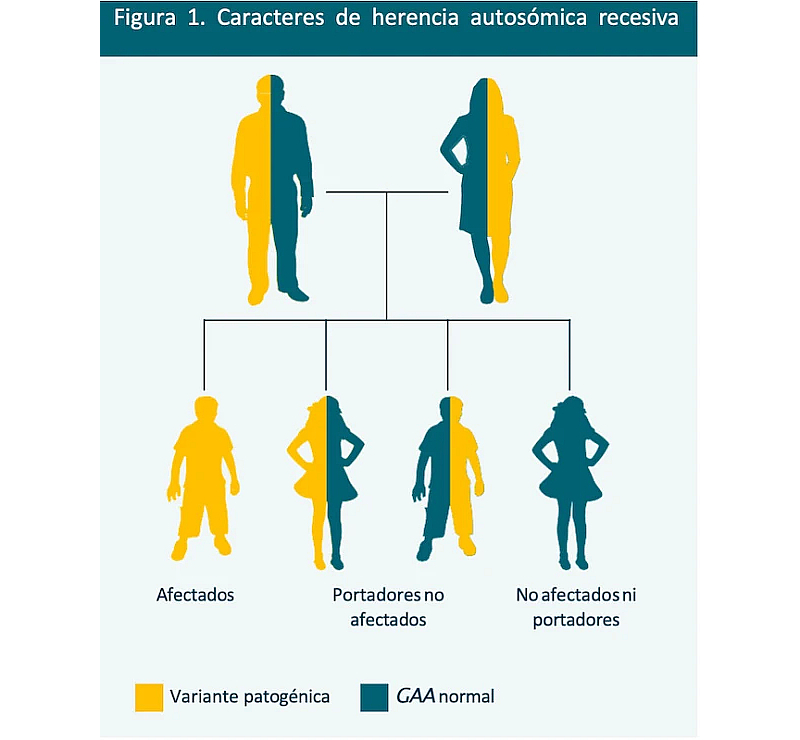
Figura adaptada de Taglia A, et al. 2015.1
La relación entre el genotipo y el fenotipo
La gravedad de la enfermedad y la edad de aparición están relacionadas con el grado de carencia enzimática;1 en general, el tipo de variante en GAA predice el fenotipo clínico;2 no obstante, hay factores como los genes modificadores y el ambiente, que pueden modular las relaciones entre el genotipo y el fenotipo, e influir en la edad de aparición, la gravedad y la evolución de la enfermedad de Pompe.3
Las variantes patogénicas pueden ser leves, polimórficas o graves (Figura 2).2 La herencia de dos variantes patogénicas graves suele traducirse en la ausencia total o casi total de actividad GAA y en la presencia de enfermedad de Pompe de inicio temprano. La herencia de una variante grave y otra leve, o de dos leves, suele traducirse en la presencia de actividad GAA residual y enfermedad de Pompe de inicio tardío.2 La presentación clínica y la evolución de la LOPD pueden variar incluso entre pacientes con el mismo genotipo, como pone de manifiesto la variabilidad intrafamiliar entre hermanos con LOPD.3
Figura 2. Relación entre las variantes, la actividad GAA y el diagnóstico
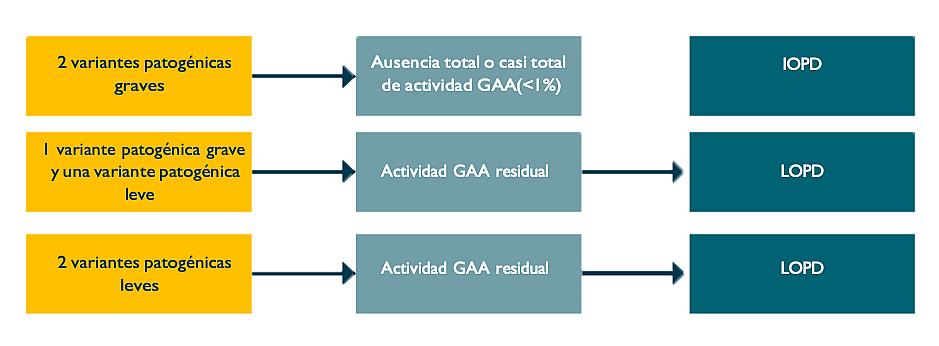
Figura adaptada de Kroos M, et al. 2012.2
Asesoramiento genético y pruebas genéticas de la enfermedad de Pompe
El diagnóstico temprano es una prioridad para los pacientes con enfermedad de Pompe porque permite iniciar el tratamiento de forma temprana y así obtener mejores resultados.3 El método diagnóstico de elección depende de los antecedentes familiares de enfermedad de Pompe (IOPD o LOPD) y de si se aplica a los familiares de primer y segundo grado, como se muestra en el siguiente cuadro.3

Tabla elaborada a partir de McIntosh P, et al. 2017.3
*El estudio enzimático no es un método fiable para determinar el estado del portador y solo puede utilizarse para detectar la enfermedad en individuos afectados.4,1
El diagnóstico temprano, basado en estas pruebas, y el tratamiento adecuado de la enfermedad son esenciales para evitar que el deterioro muscular irreversible limite la movilidad y la actividad diaria del paciente.4,1
1. Taglia A, et al. Acta Myologica 2011;30:179-181. 2. Kroos M, et al. Am J Med Genet Semin Med Genet 2012;160C:59-68. 3. McIntosh P, et al. Am J Genet 2017;173A:2628-2634. 4. Rigter T, et al. Mol Genet Metab 2012;107(3):448-455.
Manifestaciones clínicas de la enfermedad
En la mayoría de los casos, los pacientes con enfermedad de Pompe presentan signos de disfunción motora y respiratoria1,2,5
-
77% de los pacientes con LOPD presenta intolerencia al ejercicio y debilidad muscular de cinturas.
-
55% de los pacientes con LOPD presenta insuficiencia respiratoria y debilidad muscular de cinturas.
-
IOPD Los sintomas aparecen en la infancia temprana, se agudizan de forma rápida y suelen causar la muerte antes de los 2 años de edad. Las manifestaciones más frecuentes son: miocardiopatía, hipotonia, debilidad muscular, dificultad respiratoria, problemas de alimentación y retraso del crecimiento.2,3,9
-
Musculatura facial y bulbar:15
-
Dificultad para hablar
-
Dificultad para tragar
-
Debilidad enlos músculos faciales (ej. ptosis)
-
-
Aparato digestivo:2,16,17
-
Pérdida de peso sin causa aparente
-
Incontinencia fecal o urinaria
-
Molestias gastrointestinales
-
-
Aparato musculoesquelético:2,15,18-21
-
Escoliosis
-
Osteoporosis
-
Osteopenia
-
Fracturas vertebrales
-
HiperCKemia persistente sin casusa aparente (valores entre 1,5 y 15 veces por encima de los normales; ~300-2.000 U/L)
-
-
Musculatura proximal y de cinturas
-
Debilidad en las piernas2
-
Dificultad para subir escaleras22
-
Dificultad para levantarse de una silla
-
Vaivén de las caderas al caminar o marcha de pato2
-
Caídas frecuentes y problemas para correr o practicar deporte5
-
-
Musculatura respiratoria
-
Disnea de esfuerzo, por ejemplo, al subir las escaleras22,24
-
Dificultad para respirar, sobre todo en posición de decúbito2
-
Neumopatía restrictiva y tos ineficaz26
-
Dificultad respiratoria nocturna, con apnea del sueño, cefaleas matutinas o elevación del dióxido de carbono (Co2)25
-
Hipoventilación durante el sueño25
-
Cansancio diurno excesivo25
-
Intolerancia al ejercicio1
-
La LOPD se manifiesta en muchas partes del cuerpo
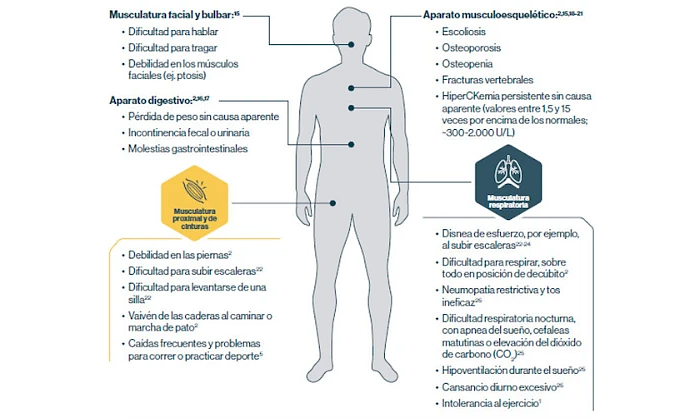
Si detecta estos signos y sintomas en algún paciente, solicite cuanto antes las pruebas diagnósticas de la enfermedad de Pompe.4
CK: creatina quinasa; IOPD: enfermedad de Pompe de inicio infantil; LOPD: enfermedad de Pompe de inicio tardío
1. Schüller A, et al. Am J Med Genet C Semin Med Genet. 2012;160C(1):80-88. 2. Kishnani PS, et al. Genet Med. 2006;8(5):267-288. 3. van Capelle CI, et al. Orphanet J Rare Dis. 2016;11(1):65. 4. Kishnani PS, et al. J Pediatr. 2004;144(5 suppl):S35-S43. 5. Kishnani PS, et al. J Pediatr. 2006;148(5):671-676. 6. Chan J, et al. Mol Genet Metab. 2017;120(3):163-172. 7. Karabul N, et al. JIMD Rep. 2014;17:53-61. 8. Bernstein DL, et al. Mol Genet Metab. 2010;101(2-3):130-133. 9. Rigter T, et al. Mol Genet Metab. 2012;107(3):448-455. 10. Toscano A, et al. Acta Myol. 2013;32(2):78-81. 11. Preisler N, et al. Mol Genet Metab. 2013;110(3):287-289. 12. Manganelli F, et al. Acta Myol. 2013;32(2):82-84. 13. Moghadam-Kia S, et al. Cleve Clin J Med. 2016;83(1):37-42. 14. Mellies U, et al. Respir Med. 2009;103(4):477-484. 15. Boentert M, et al. Int J Mol Sci. 2016;17(10):1735. 16. Fuller DD, et al. Respir Physiol Neurobiol. 2013;189(2):241-249. 17. American Association of Neuromuscular and Electrodiagnostic Medicine. Muscle Nerve. 2009;40(1):149-160.
Desafíos cotidianos
Entre otras manifestaciones clínicas, la LOPD provoca una afectación musculoesquelética. Consulta las dificultades más características de los pacientes con Enfermedad de Pompe.
Historia natural de la enfermedad de Pompe
No existen estudios prospectivos en los que se haya hecho seguimiento a pacientes presintomáticos con la enfermedad de Pompe para saber en qué momento se inician los síntomas. Los estudios de los que disponemos en en este sentido son retrospectivos. El grupo del Dr. Hagemans revisó la historia clínica de 54 pacientes y evidenció que en la mayoría de los pacientes los primeros síntomas comienzan en la tercera década de la vida (Figura 1), normalmente con debilidad proximal de las extremidades inferiores, que se manifiesta en forma de alteración de la marcha o dificultad para correr, subir escaleras o para levantarse de la silla.1
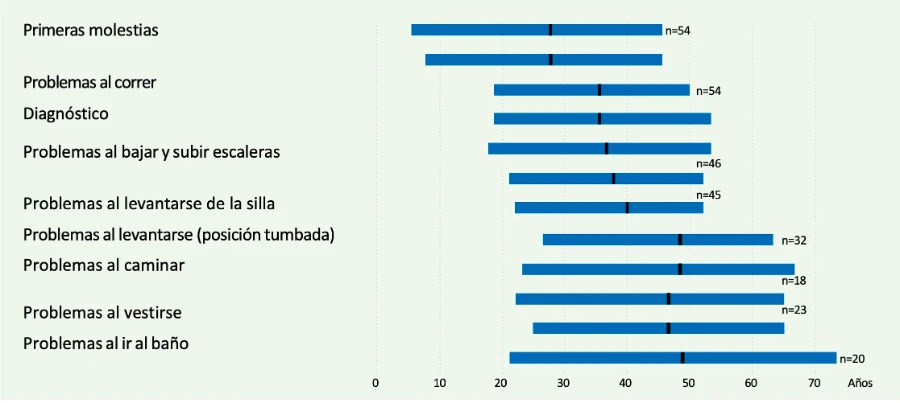
Figura 1. Inicio de la enfermedad y progresión clínica en pacientes con enfermedad de Pompe del adulto (Hagemans ML, et al. Brain 20051).
Como se puede ver en la Figura 1, existe una progresión clínica continua desde el inicio de los síntomas, que se caracteriza por un deterioro de las habilidades motoras, lo que lleva al paciente a tener una discapacidad importante, precisando de silla de ruedas entre los 40 y 50 años de media. La mayoría de los pacientes desarrolla alteraciones respiratorias en algún momento de la enfermedad, lo que supone la necesidad de ventilación nocturna no invasiva, generalmente a partir de los 50 años.1
Existen estudios prospectivos que han analizado la pérdida de fuerza muscular en un año de vida natural de pacientes con enfermedad de Pompe del adulto, bien a través del balance muscular manual, bien usando miómetros de mano o fijos de pared.2,3 Existe una correlación positiva entre la duración de la enfermedad y el grado de debilidad de los pacientes (Figuras 2A y 2B).
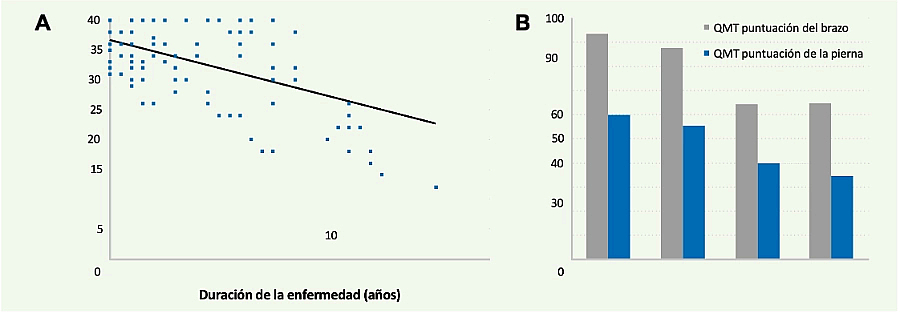
Figura 2. Correlación positiva entre el tiempo de evolución de la enfermedad y el grado de debilidad muscular de los pacientes, evaluado de forma manual (balance muscular) (A) o mediante el uso de miómetros (B) (Van der Beek NA, et al. Neuromuscular disorders 20092 (A) y Wokke JH, et al. Muscle & Nerve 20083 (B)).
Se han realizado estudios prospectivos de un año de seguimiento en pacientes con diversos grados de enfermedad de Pompe.3 Estos estudios demostraron una pérdida progresiva de fuerza muscular, la cual en algunos grupos musculares como el cuádriceps o el bíceps podía ser cercana al 10% respecto al inicio del seguimiento (Figura 3).
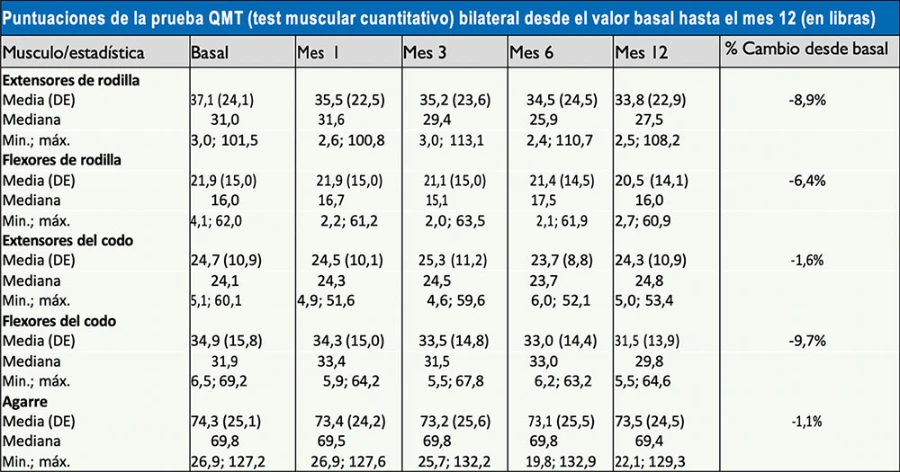
Figura 3. Estudio con miómetro de la fuerza muscular de un grupo de pacientes con enfermedad de Pompe del adulto durante un año. Este estudio demostró una pérdida progresiva de fuerza muscular, especialmente en los músculos proximales de las extremidades superiores e inferiores (Wokke JH, et al. Muscle & Nerve 20083). DE: desviación estándar.
De forma paralela a lo que ocurre en el músculo esquelético, se ha podido demostrar un empeoramiento progresivo de la función ventilatoria en los pacientes con enfermedad de Pompe del adulto.2,3 Este deterioro es debido a una debilidad progresiva de la musculatura diafragmática. Existe una correlación negativa entre el tiempo de duración de la enfermedad y la capacidad vital forzada (CVF) residual (Figura 4). Se estima que la pérdida anual de CVF es del 4,6%.3
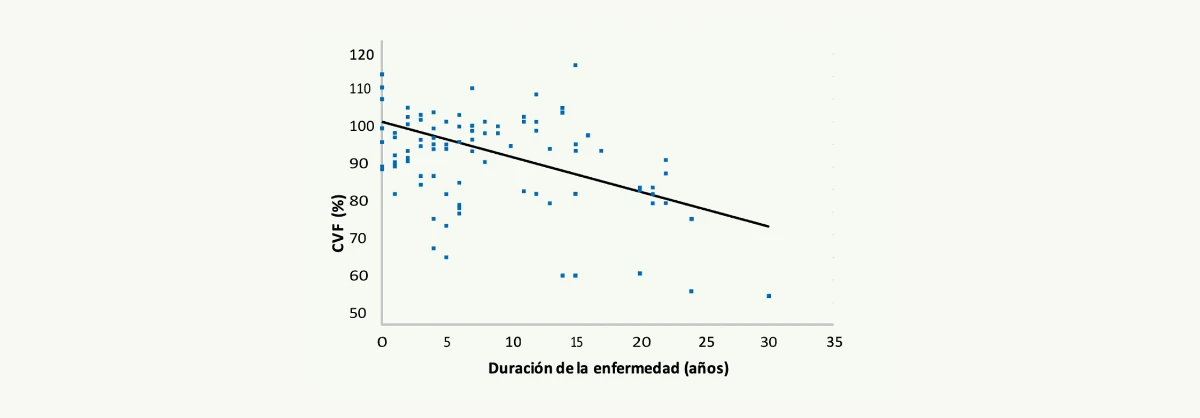
Figura 4. Correlación entre el tiempo de evolución de la enfermedad y la capacidad vital forzada de los pacientes con enfermedad de Pompe del adulto (Van der Beek NA, et al. Neuromuscular Disorders 20092).
Este empeoramiento progresivo en la fuerza del músculo esquelético y en la capacidad ventilatoria lleva a un grado de discapacidad importante. Los pacientes deben abandonar actividades físicas, siendo frecuente el uso de silla de ruedas para poder desplazarse. Asimismo, es preciso el uso de ventilación invasiva debido a la disfunción ventilatoria.2
Además, la enfermedad de Pompe se asocia a un mayor riesgo de mortalidad. En un estudio publicado en 20134, de un grupo de 268 pacientes adultos, 34 fallecieron durante el período de seguimiento. Los factores relacionados con el riesgo de mortalidad eran el tiempo de evolución de la enfermedad y el grado de discapacidad física en el momento del diagnóstico (Figura 5).4
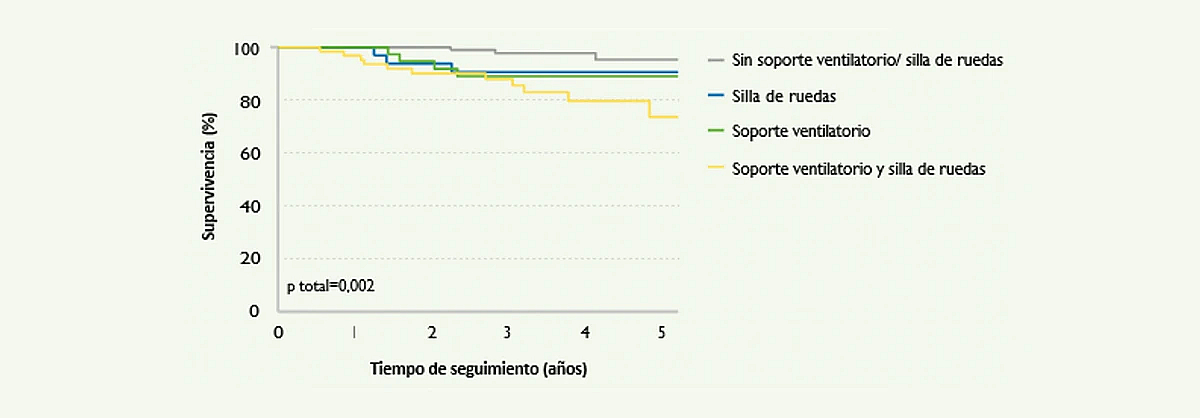
Figura 5. Curvas de Kaplan Meier de estimación del riesgo de mortalidad en una cohorte de 268 pacientes con enfermedad de Pompe. Se puede observar una buena correlación entre el grado de mortalidad y la necesidad de soporte ventilatorio y/o uso de silla de ruedas (Güngör D, et al. Orphanet Journal of Rare Diseases 20134).
En resumen, los estudios realizados han mostrado que los pacientes con enfermedad de Pompe desarrollan debilidad muscular continua que afecta a las extremidades superiores e inferiores y a la musculatura ventilatoria. Como consecuencia de esta debilidad, precisan del uso de silla de ruedas y aparatos de ventilación no invasiva. La enfermedad de Pompe supone un aumento del riesgo de mortalidad en los pacientes, siendo el grado de discapacidad en el momento de inicio del seguimiento el factor de riesgo más importante para fallecer en un período de tiempo de 5 años.2-4
1. Hagemans ML, Winkel LP, van Doorn PA, et al. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain. 2005;128(Pt 3):671-7. 2. Van der Beek NA, Hagemans ML, Reuser AJ, et al. Rate of disease progression during long-term follow-up of patients with late-onset Pompe disease. Neuromuscul Disord. 2009;19(2):113-7. 3.Wokke JH, Escolar DM, Pestronk A, et al. Clinical features of late-onset Pompe disease: a prospective cohort study. Muscle Nerve. 2008;38(4):1236-45. 4. Güngör D, Kruijshaar ME, Plug I, et al. Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J Rare Dis. 2013;8:49.
Diagnóstico diferencial
La capacidad de diferenciar correctamente la enfermedad de Pompe de otras enfermedades es crucial para minimizar el retraso en el diagnóstico y optimizar los desenlaces de los pacientes.
Sin embargo, dado que la enfermedad de Pompe es rara y comparte muchos de sus signos y síntomas con otras enfermedades, el diagnóstico diferencial puede suponer un reto.
La enfermedad de Pompe debería valorarse cuando los signos y síntomas indiquen una degeneración muscular progresiva, en lactantes, niños adolescentes y adultos:
En lactantes: cardiomegalia /cardiomiopatía, hipotonía, rápida progresión de la debilidad muscular tanto del musculo ventilatorio como del esquelético, dificultades para alimentarse, y retraso del crecimiento.1,2,3,4
En niños, adolescentes y adultos: debilidad proximal de cinturas y/o de los músculos respitatorios además de escoliosis en adolescentes y elevación de los niveles de creatinquinasa (CK).5,6
Una vez surja la sospecha clínica, puede medirse la actividad de la enzima GAA para confirmar la enfermedad de Pompe.5,6
Considere la enfermedad de Pompe en el diagnóstico diferencial de estas otras enfermedades:1
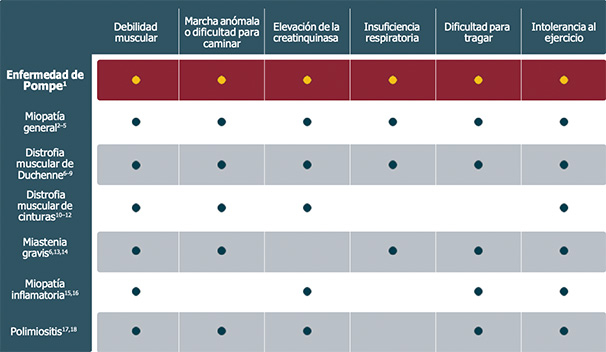.2024-05-06-14-51-08.jpg)
Poblaciones de alto riesgo
Como se muestra en las siguientes tablas, existen datos convincentes que respaldan la realización de pruebas rutinarias para detectar la enfermedad de Pompe (a partir de un análisis para detectar la enzima GAA) en poblaciones de pacientes que presentan ciertos síntomas sugestivos de la enfermedad de Pompe, tales como debilidad de cinturas y/o niveles elevados de creatinquinasa (CK), con o sin la debilidad de los músculos respiratorios:
Diagnósticos de la enfermedad de Pompe en pacientes con debilidad de cinturas.7,8,9,10
|
Autor |
Criterios para realizar pruebas |
Número de pacientes que se han sometido a pruebas |
Número de pacientes con enfermedad de Pompe |
|
Goldstein (2009) |
Paciente adulto con debilidad muscular de la cintura o de las extremidades proximales y sospecha de enfermedad Pompe |
132 |
207132(15%) |
|
Preisler (2013) |
Distrofia muscular de la cintura y extremidades sin clasificar |
38 |
3/38 (8%) |
|
Willis (2012) |
Debilidad muscular de la cintura y extremidades ± síntomas respiratorios ± escoliosis ± columna vertebral rígida |
102 |
5/102 (4,9%) |
| Bautista lorite (2013) | Debilidad muscular de la cintura y extremidades | 144 | 15/144(10,2%) |
Diagnósticos de la enfermedad de Pompe en pacientes con creatina-cinasa (CK) elevada10,11,12
|
Autor |
Criterios para realizar pruebas |
Número de pacientes que se han sometido a pruebas |
Número de pacientes con enfermedad de Pompe |
|
Bautista lorite (2013) |
HiperCKmias idiopáticas |
205 |
5/205(2,4%) |
|
Fernandez (2006) |
HiperCKmias oligosintomática o asintomática idiopática |
104 |
4/104 (3,8%) |
|
Spada (2013) |
Pacientes con HiperCKemia asintomática idiopática |
137 |
3/137(2,2%) |
1. Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8:267-88. 2. Gilbert-Barness E. Review: Metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 2004; 34:15-34. 3. Howell RR, Byrne B, Darras BT, Kishnani P, Nicolino M, van der Ploeg A. Diagnostic challenges for Pompe disease: An under-recognized cause of floppy baby syndrome. Genet Med 2006:8;1-8. 4. Gilchrist JM. Overview of neuromuscular disorders affecting respiratory function. Semin Respir Crit Care Med 2002; 23:191-200. 5. American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009;40:149-60. 6. Ausems MG, Lochman P, van Diggelen OP, Ploos van Amstel HK, Reuser AJ, Wokke JH. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology. 1999 Mar 10;52(4):851-3. 7. Goldstein JL, Young SP, Changela M, Dickerson GH, Zhang H, Dai J, Peterson D, Millington DS, Kishnani PS, Bali DS (2009). Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve 40:32-36. 8. Preisler N, Lukacs Z, Vinge L, Madsen KL, Husu E, Hansen RS, Duno M, Andersen H, Laub M, Vissing J (2013). Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol Genet Metab 110(3):287-9. 9. Willis T, Roberts M, Hilton-Jones D, Quinlivan R,5 Hanna M, Straub V (2012). Detection rate of Pompe disease in undiagnosed neuromuscular patients from four major centre’s in the UK- Results of a 12 month prospective audit. BMC Musculoskelet Disord 14(Suppl 2):P20. 10. Bautista Lorite J (2013) Detección de la enfermedad de Pompe en pacientes con distrofia de cinturas indefinidas o hiperCKemias asintomáticas. Expert Rev Neur Ed especial Octubre 2013:17-19. 11. Fernandez C, de Paula AM, Figarella-Branger D, Krahn M, Giorgi R, Chabrol B, et al. (2006). Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 66:1585-7. 12. Spada M, Porta F, Vercelli L, Pagliardini V, Chiadò-Piat L, Boffi P, Pagliardini S, Remiche G, Ronchi D, Comi G, Mongini T (2013). Screening for later-onset Pompe's disease in patients with paucisymptomatic hyperCKemia. Mol Genet Metab 109:171-173.
Pruebas diagnósticas y de confirmación
La enfermedad de Pompe es una enfermedad rara con signos y síntomas similares a los de otras muchas enfermedades.
Esto significa que la enfermedad de Pompe tiende a pasar desapercibida, al menos al principio, hasta que se han descartado otras enfermedades más frecuentes, lo que da como resultado un retraso en el diagnóstico que puede ser perjudicial o incluso potencialmente mortal.
En lactantes, el diagnóstico temprano es especialmente importante, ya que la falta de tratamiento suele conllevar la muerte durante el primer año de vida. Sin embargo, se ha observado que los lactantes experimentan una mediana de retraso de 2,7 meses desde la primera aparición de los signos hasta el diagnóstico de la enfermedad de Pompe.1
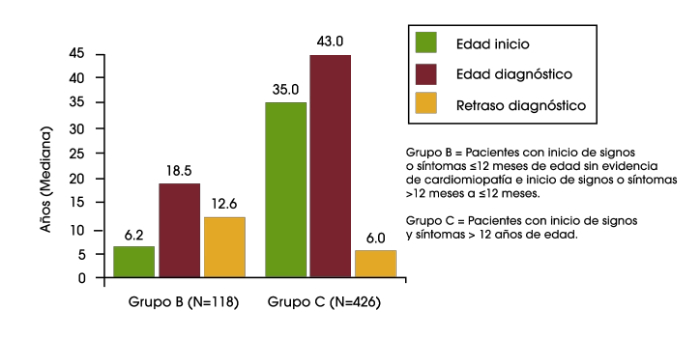
En un análisis reciente de los datos del Registro de Pompe se encontró retrasos diagnósticos en lactantes, niños y adolescentes/adultos. En los bebés que tuvieron el inicio de los síntomas durante los primeros 12 meses de vida, y que presentaban cardiomiopatía (es decir, la enfermedad de Pompe infantil clásica), el retraso diagnóstico medio fue de 1,4 meses. Para los pacientes con inicio de síntomas sobre la edad de 12 años, el retraso diagnóstico medio fue de 6,0 años. El retraso más largo en el diagnóstico fue una media de 12,6 años, que se encontró en los pacientes que tuvieron inicio de los síntomas durante los primeros 12 meses de vida, pero no tenían cardiomiopatía, o tuvieron el inicio de los síntomas entre los 12 meses y 12 años de edad.2
Por tanto, existe la necesidad de un diagnóstico precoz en todo el espectro de la enfermedad de Pompe, que ayude a optimizar los resultados del paciente.
Retraso del diagnósitco en lactantes, niños y adolescentes/ adultos con la enfermedad de Pompe2
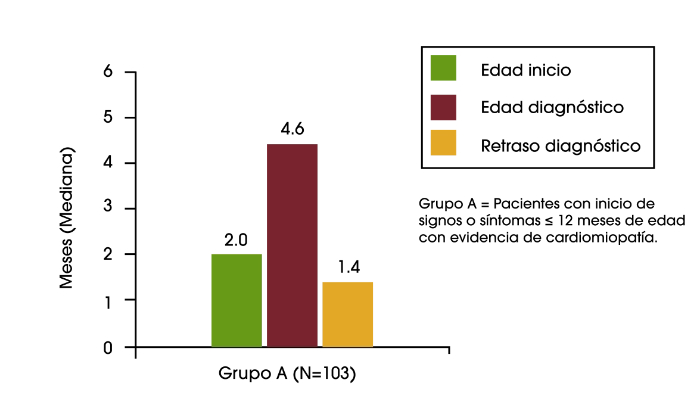.2024-05-06-14-51-08.jpg)
.2024-05-06-14-51-08.jpg)
La relación entre el genotipo y el fenotipo
La gravedad de la enfermedad y la edad de aparición están relacionadas con el grado de carencia enzimática;1 en general, el tipo de variante en GAA predice el fenotipo clínico;2 no obstante, hay factores como los genes modificadores y el ambiente, que pueden modular las relaciones entre el genotipo y el fenotipo, e influir en la edad de aparición, la gravedad y la evolución de la enfermedad de Pompe.3
Las variantes patogénicas pueden ser leves, polimórficas o graves (Figura 2).2 La herencia de dos variantes patogénicas graves suele traducirse en la ausencia total o casi total de actividad GAA y en la presencia de enfermedad de Pompe de inicio temprano. La herencia de una variante grave y otra leve, o de dos leves, suele traducirse en la presencia de actividad GAA residual y enfermedad de Pompe de inicio tardío.2 La presentación clínica y la evolución de la LOPD pueden variar incluso entre pacientes con el mismo genotipo, como pone de manifiesto la variabilidad intrafamiliar entre hermanos con LOPD.3
Diagnóstico
Aunque las vías clínicas para el diagnóstico de la enfermedad de Pompe son variables, el proceso implica generalmente:
Evaluación clínica de los síntomas presentes por un médico de atención primaria.
Remisión a un especialista para una investigación clínica en mayor profundidad, incluyendo pruebas analíticas o clínicas adicionales.
Pruebas de confirmación.
Diagnóstico definitivo
La enfermedad de Pompe se confirma por una ausencia completa o una disminución marcada de la actividad de la alfa glucosidasa ácida (GAA).3,4 La actividad residual de la GAA en pacientes con enfermedad de Pompe puede oscilar entre menos del 1 % (generalmente en lactantes) y el 40% de los niveles normales.5 La enfermedad de Pompe también puede confirmarse mediante el análisis de mutaciones, que muestra la presencia de dos alelos mutados.
Tradicionalmente, la prueba para detectar la enzima GAA se realizaba a partir de un cultivo de fibroblastos cutáneos.3,4 Sin embargo, la recogida de muestras es relativamente invasiva y transcurren aproximadamente 6 semanas hasta obtener los resultados. Se desaconseja claramente este prolongado período de espera, especialmente en lactantes con una enfermedad que progresa con rapidez. Por tanto, el uso de muestras de sangre, incluidas las muestras de sangre seca, se está convirtiendo en la práctica habitual. La extracción de muestras de sangre para la confirmación de la enfermedad de Pompe es mínimamente invasiva, exacta y puede generalmente ofrecer resultados en tan solo unos días.3,4
Si se encuentra una actividad enzimática reducida de GAA, se debe confirmar con una segunda muestra o por análisis genético.6
Aunque las biopsias musculares son una opción para las pruebas de actividad de la GAA, no suelen ser la opción preferida, ya que son invasivas y tienen un alto riesgo de falsos positivos si se manipulan las muestras de forma incorrecta. Las biopsias musculares pueden ser útiles para la evaluación histológica, pero es importante tener en cuenta que el contenido de glucógeno puede variar ampliamente entre los músculos, por lo que las biopsias aparentemente normales no descartan la enfermedad de Pompe.4 Así pues, un diagnóstico de la enfermedad de Pompe debe confirmarse siempre por la ausencia o reducción de la actividad de la GAA, o por un análisis genético.
1. Kishnani PS, Hwu W-L, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006; 148:671-676. 2. Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J; Pompe Registry Boards of Advisors. Timing of diagnosis of patients with Pompe disease: data from the Pompe registry. Am J Med Genet A. 2013;161A(10):2431-43. 3. Zhang H, Kallwass H, Young SP, et al. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet med 2006; 8:302-306. 4. Winchester B, Bali D, Bodamer OA, et al for The Pompe Disease Diagnostic Working Group. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008;93(3):275-281. 5. Van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372(9646):1342-53 6. American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009;40:149-60.
Guías para el tratamiento de la enfermedad de Pompe
Aunque la base subyacente a la enfermedad de Pompe es la degeneración muscular progresiva, la enfermedad puede afectar a diferentes órganos y sistemas. Por tanto, quien mejor puede gestionar la atención al paciente y el tratamiento de este trastorno multisistémico es un equipo multidisciplinario de profesionales sanitarios1.
Manejo de la Enfermedad
Tratamiento y guías
La enfermedad de Pompe tiene tratamiento aprobado en muchos países. Este tratamiento es la enzima alglucosidasa alfa (también conocida como Myozyme).
Myozyme® se usa para tratar a adultos, niños y adolescentes de todas las edades con diagnóstico confirmado de la enfermedad de Pompe. Myozyme reemplaza a la enzima natural que falta en la enfermedad de Pompe.
Contenido mínimo de Myozyme®
PRESENTACIÓN, PRECIO Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN:
Myozyme 50 mg polvo concentrado para solución para perfusión – 1 vial (CN 654213.2): PVP: 580,91 €, PVP IVA: 604,15 €. Medicamento sujeto a prescripción médica. Financiado por el SNS. Uso hospitalario.

¿Tienes alguna pregunta? Escríbenos para resolver tus dudas.
Consulta de Información Médica
MAT-ES-2301236 V2 Septiembre 2025