- Artículo
- Fuente: Campus Sanofi
- 10 jul 2024
¿Qué es el ASMD?
ASMD: unas siglas que pueden cambiar tu enfoque diagnóstico.
Es una enfermedad de depósito lisosomal causada por el déficit de esfingomielinasa ácida1.
Puede presentarse en la infancia o en la edad adulta2.
Cursa con hepatoesplenomegalia, trombocitopenia y afectación pulmonar, entre otras manifestaciones¹.
Hay distintos tipos: A, AB y B3.
Todos con riesgo de daño progresivo si no se identifican2.
Conoce más sobre el ASMD y mejora tu capacidad de detección con los contenidos que ves en pantalla.
ASMD: unas siglas que pueden cambiar tu enfoque diagnóstico.
Es una enfermedad de depósito lisosomal causada por el déficit de esfingomielinasa ácida1.
Puede presentarse en la infancia o en la edad adulta2.
Cursa con hepatoesplenomegalia, trombocitopenia y afectación pulmonar, entre otras manifestaciones¹.
Hay distintos tipos: A, AB y B3.
Todos con riesgo de daño progresivo si no se identifican2.
Conoce más sobre el ASMD y mejora tu capacidad de detección con los contenidos que ves en pantalla.
Continúa navegando para descifrar el ASMD junto a GlorIA:
Características de ASMD
El Déficit de Esfingomielinasa Ácida (ASMD) es una enfermedad genética grave y progresiva1.
También conocida como enfermedad de Niemann-Pick A, A/B y B, el ASMD es una enfermedad genética de depósito lisosomal que afecta tanto a pacientes pediátricos como adultos1.
- El ASMD es una enfermedad progresiva y potencialmente mortal causada por un déficit en la actividad enzimática1.
- Su aparición y progresión son heterogéneas y puede conducir a complicaciones multiorgánicas1.
- Los signos y síntomas se suelen manifestar en el bazo, hígado, pulmones y sistema hematológico; en algunos tipos de la enfermedad, el sistema nervioso también se ve afectado1.
Espectro de ASMD
ASMD cuenta con un amplio espectro de la enfermedad, que se divide en 3 tipos1
|
Tipo A Progresa rápidamente con manifestaciones multiorgánicas agudas y neurodegeneración1 |
Tipo A/B Progresa de forma variable con manifestaciones multiorgánicas y distintos grados de afección neurológica1 |
Tipo B Crónica con manifestaciones multiorgánicas y sin afección o afección neurológica menor1 |
|
Inicio Infancia temprana1 |
Inicio Infancia a niñez1,2 |
Inicio Infancia a fase adulta2 |
|
Esperanza de vida 2-3 años de edad1 |
Esperanza de vida Entre niñez temprana y fase adulta1 |
Esperanza de vida Entre niñez y fase adulta tardía1 |
Fisiopatología de ASMD
Todos los tipos de ASMD responden a una misma causa: déficit enzimático1
La deficiencia de la enzima esfingomielinasa ácida (ASM) conlleva a una acumulación intracelular de esfingomielina que resulta en daños multiorgánicos progesivos1
- ASMD es una enfermedad de depósito lisosomal con herencia autosómica recesiva provocada por variantes patogénicas del gen codificante para la enzima ASM, el gen de la esfingomielina fosfiesterasa 1 (SMPD1)1
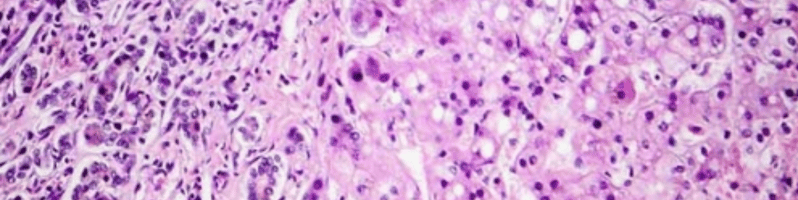
Núcleo, lisosoma y esfingomielina
- La esfingomielina favorece la regulación de procesos celulares tales como el ciclo celular, la señalización celular y la apoptosis3-5.
- La enzima ASM descompone las moléculas de esfingomielina, catalizando su hidrólisis a ceramida y fosfocolina en los lisosomas3.
.2024-04-22-14-59-54.jpg)
Examen histológico de un macrófago en un paciente con ASMD.
Imagen empleada con el consentimiento de Jesús Villarubia, MD, PhD, Hospital Universitario Ramón y Cajal de Madrid, España.
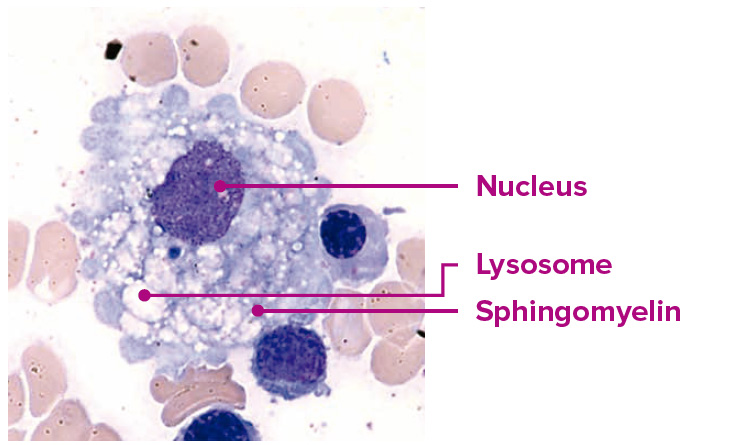
Núcleo, esfingomielina
- Cuando la actividad de la enzima ASM es deficiente, la esfingomielina no puede ser metabolizada correctamente, lo que provoca su acumulación en los lisosomas, particularmente en los macrófagos y hepatocitos1.
- Esta acumulación daña a las células y a múltiples órganos, lo que conlleva a complicaciones potencialmente mortales1.
Examen histológico de un macrófago en un paciente con ASMD.
Imagen empleada con el consentimiento de Jesús Villarubia, MD, PhD, Hospital Universitario Ramón y Cajal de Madrid, España.
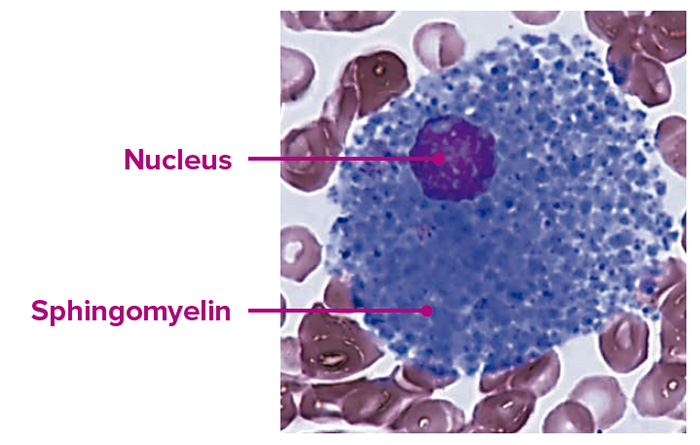
Núcleo, lisosoma y esfingomielina
- La esfingomielina favorece la regulación de procesos celulares tales como el ciclo celular, la señalización celular y la apoptosis3-5.
- La enzima ASM descompone las moléculas de esfingomielina, catalizando su hidrólisis a ceramida y fosfocolina en los lisosomas3.
Examen histológico de un macrófago en un paciente con ASMD.
Imagen empleada con el consentimiento de Jesús Villarubia, MD, PhD, Hospital Universitario Ramón y Cajal de Madrid, España.
Núcleo, esfingomielina
- Cuando la actividad de la enzima ASM es deficiente, la esfingomielina no puede ser metabolizada correctamente, lo que provoca su acumulación en los lisosomas, particularmente en los macrófagos y hepatocitos1.
- Esta acumulación daña a las células y a múltiples órganos, lo que conlleva a complicaciones potencialmente mortales1.
Examen histológico de un macrófago en un paciente con ASMD.
Imagen empleada con el consentimiento de Jesús Villarubia, MD, PhD, Hospital Universitario Ramón y Cajal de Madrid, España.

Sospechas y derivación
Todos los tipos de ASMD pueden provocar graves consecuencias
Independientemente de dónde se sitúan los pacientes dentro del espectro de la enfermedad, ASMD puede tener un impacto negativo en su bienestar y calidad de vida.
Reconozca los signos y síntomas clave para identificar de forma temprana ASMD1
Continúa explorando otros contenidos:
.png)
Presentación Clínica y diagnóstico ASMD
El ASMD se presenta con síntomas heterogéneos y progresivos
.png)
Fisiopatología y clasificación ASMD
Descubre más con GlorIA

Solicitud DBS y Contacto ASMD
Sospechar ASMD puede cambiar una vida
.png)
¿ASMD o GAUCHER?
Descubre las diferencias entre estas dos enfermedades
Presentación Clínica y diagnóstico ASMD
El ASMD se presenta con síntomas heterogéneos y progresivos
Fisiopatología y clasificación ASMD
Descubre más con GlorIA
Solicitud DBS y Contacto ASMD
Sospechar ASMD puede cambiar una vida
¿ASMD o GAUCHER?
Descubre las diferencias entre estas dos enfermedades


Referencias
- McGovern MM, Avetisyan R, Sanson BJ, et al. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. 2017;12(1):41.
- Cassiman D, Packman S, Bembi B, et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases [published correction appears in Mol Genet Metab. 2018 Dec;125(4):360]. Mol Genet Metab. 2016;118(3):206-213.
- Schuchman EH, Desnick RJ. Types A and B Niemann-Pick disease. Mol Genet Metab. 2017;120(1-2):27-33.
- Eyster KM. The membrane and lipids as integral participants in signal transduction: lipid signal transduction for the non-lipid biochemist. Adv Physiol Educ. 2007;31(1):5-16.
- Lee S, Lee YS, Choi KM, et al. Quantitative analysis of sphingomyelin by high-performance liquid chromatography after enzymatic hydrolysis. Evid Based Complement Alternat Med. 2012;2012:396218.
MAT-ES-2502916 V1 Septiembre 2025